Your FDA IDE approval letter is signed, the enrollment window is set, and the electronic data capture (EDC) platform you're evaluating still wants to call a device malfunction a "dosing event." For sponsors and CROs running an early feasibility or pivotal study under an Investigational Device Exemption (IDE), Viedoc's EDC software is built around device workflows from the ground up, with study builds averaging 10 weeks and a 100% FDA inspection pass rate across 8,000-plus studies. This comparison reviews five EDC platforms for IDE studies across device-specific design, in-house configuration, accountability tracking, and FDA compliance readiness.
An IDE study rarely holds still. An early feasibility study can pivot into a redesigned pivotal protocol after an interim safety review, and your clinical affairs team, often three or four people wearing multiple hats, has to absorb that change without waiting weeks for a vendor programmer. You're also tracking things a pharma-built EDC was never designed for: serial numbers, lots, and usability endpoints tied to a unit rather than a dose.
Enterprise EDC platforms built for large pharma Phase III programs bring configuration cycles calibrated for hundred-site drug trials, not a 15-site pivotal device study run by a lean team. Platforms retrofitted from drug trial terminology force you to work around fields built for "dosing," adding friction where an FDA reviewer expects clarity. The platforms below are judged on whether they fit an IDE study's actual shape: device-native workflows, in-house control, and audit trail rigor that holds up to scrutiny from FDA's Center for Devices and Radiological Health (CDRH).
Best EDC platforms for medical device IDE studies: quick comparison
| Platform | Product / module | Overview |
|---|---|---|
| Viedoc | EDC software | No-code EDC with device accountability fields, builds averaging 10 weeks, and a 100% FDA inspection pass rate across 8,000-plus studies. |
| Medidata | Rave Lite | A focused version of Medidata's Rave EDC tailored for Phase I, Phase IV, and device feasibility and post-market studies. |
| Veeva | Veeva EDC (MedTech) | EDC built for device and diagnostic trial complexity, with SDV, protocol deviation tracking, and no-downtime amendments. |
| Castor EDC | Castor EDC | No-code, API-first EDC used by device companies for pre-market clinical investigations and post-market clinical follow-up studies. |
| Medrio | Medrio CDMS/EDC | No-code EDC and data management platform supporting device studies from early feasibility through pivotal and post-market trials. |
These five eClinical platforms represent the most evaluated options for medical device IDE studies, reviewed across device-specific design, in-house configuration, accountability tracking, and inspection readiness.
1. Viedoc
Viedoc's EDC software is built for device workflows rather than retrofitted from pharma templates, with fields for device terminology, procedural visits, and usability testing instead of dosing logic that doesn't apply to a device study. Studies build in an average of 10 weeks, and self-service customers configuring straightforward designs can have a database live in as little as one day. That matters most when an early feasibility study needs to pivot into a pivotal design after an interim review, and you can't wait on a vendor queue to make the change.
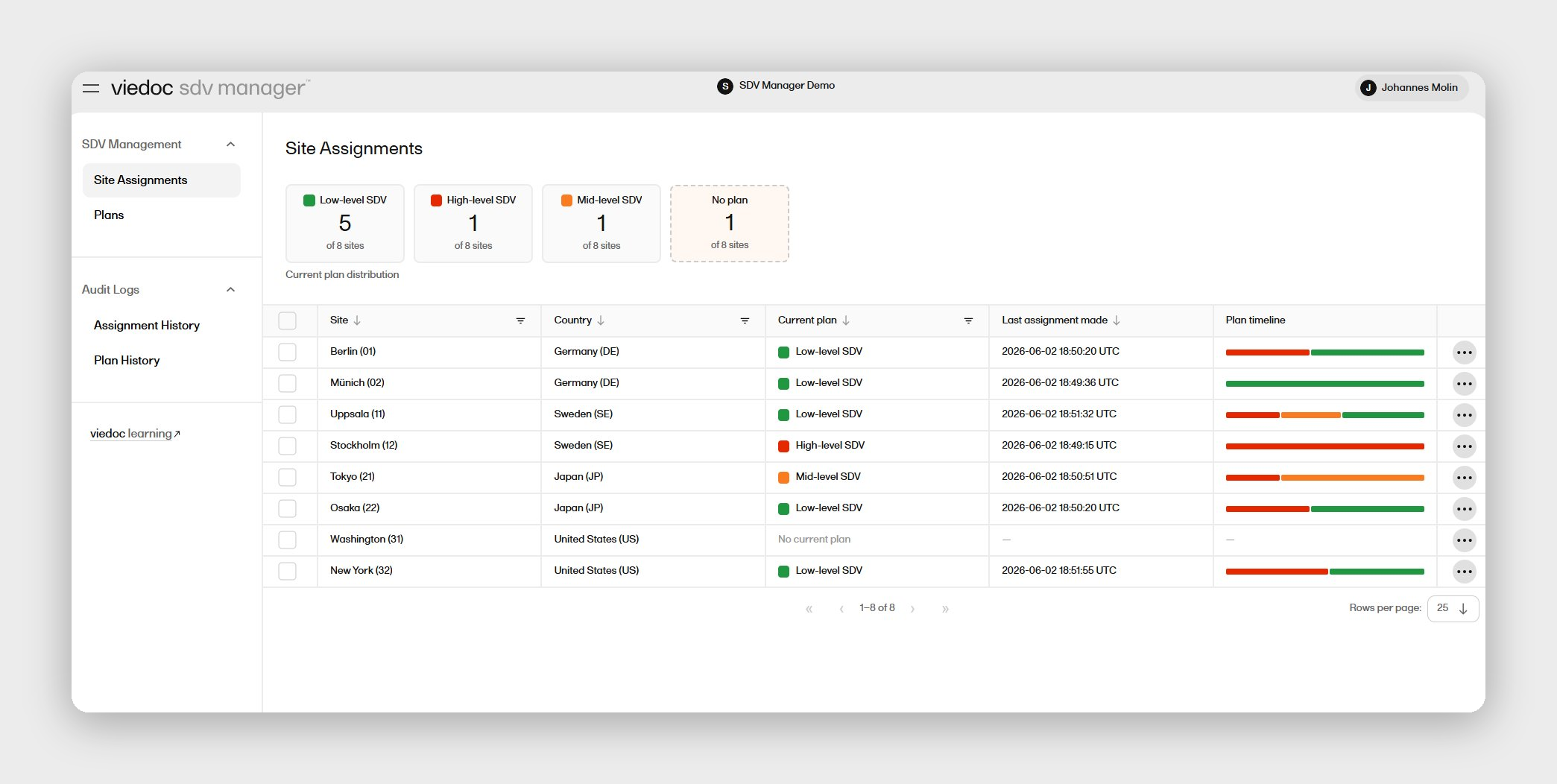
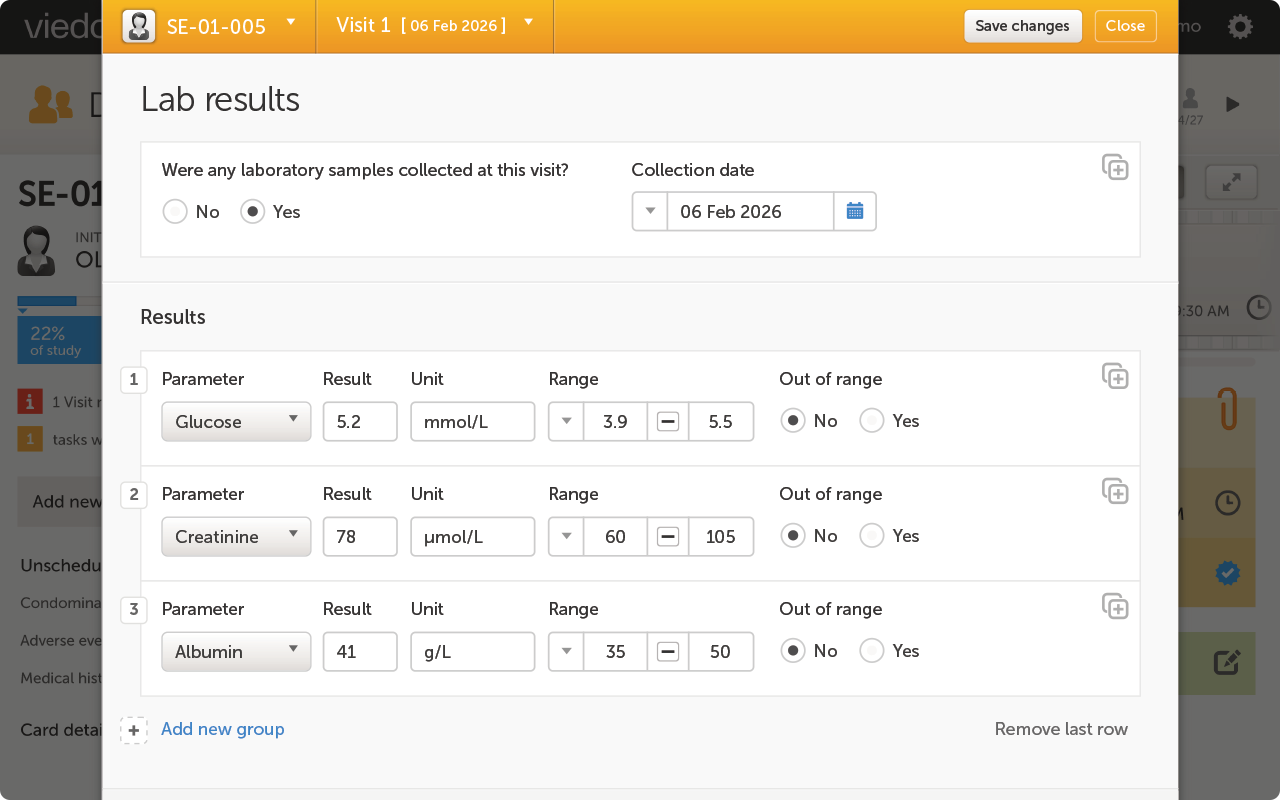
For lean clinical affairs and QA teams, the no-code Designer means your own staff configures and amends the electronic case report form (eCRF) without a vendor programmer, and configurable fields track serial numbers, lot numbers, malfunctions, and device-related events natively. Viedoc also supports trial supply management for tracking device units through replacements and returns, and standardizes device event terminology through medical coding software so every site records the same language.
On compliance, Viedoc is ISO 27001 and SOC 2 Type II certified and supports FDA 21 CFR Part 11, ICH GCP, GDPR, and HIPAA, hosted on Microsoft Azure with 99.99% uptime. Viedoc carries a 100% FDA inspection pass rate, backed by the Viedoc Inspection Readiness Packet (VIRP) at no additional cost, with 24/7 support across global offices.
Qmed, a full-service medical device CRO, chose Viedoc partly because its flexibility and reporting fit both client needs and FDA and EMA regulatory expectations across multiple device studies. "It offers customizable study workflow, and tailoring it according to trial specification is easy." – Sanchita V., Data Manager.
Verified proof points:
- Study scale: 8,000+ global studies, with 1,200-plus live trials currently powered by Viedoc's EDC
- Build speed: Study builds average 10 weeks; self-service setups possible in as little as one day
- Inspection readiness: 100% FDA inspection pass rate; VIRP included at no additional cost
- Compliance: FDA 21 CFR Part 11, ICH GCP, GDPR, HIPAA; ISO 27001 and SOC 2 Type II certified
- Device workflows: Configurable fields for serial numbers, lots, malfunctions, and device-related events
- Support: 24/7 support across global offices; unlimited user seats with transparent, study-based licensing
2. Medidata
Medidata offers Rave Lite, a focused version of its Rave EDC platform tailored specifically for Phase I, Phase IV, and medical device feasibility and post-market studies. Rave Lite runs on the same underlying technology as full Rave EDC but is scoped for smaller, right-sized studies, with pre-configured, pre-validated components intended to accelerate study builds. Medidata is a Dassault Systemes company, and its broader Rave EDC platform has been used across more than 30,000 studies with over 9 million patients and volunteers, spanning Phase I through Phase IV drug and device programs. For device sponsors running a single early feasibility or post-market study, Rave Lite offers a narrower feature set and pricing model than Medidata's full enterprise platform, which is built for large, multi-region pharma programs.
3. Veeva
Veeva offers Veeva EDC as part of its MedTech clinical data suite, positioned for device and diagnostic trials that involve patient and sample-level data beyond a standard drug protocol. The platform is used to design case report forms and edit checks without custom programming, and it includes quality controls for querying, targeted source data verification (SDV), and protocol deviation tracking. Protocol amendments deploy to the Veeva EDC database without downtime, and the EDC module connects to Veeva's broader Vault Clinical Suite, which includes CTMS, eTMF, RTSM, and eCOA on a single cloud platform. Veeva also offers a MedTech-specific Regulatory Information Management (RIM) Platform for device and diagnostics manufacturers to manage registrations and submissions. Veeva's clinical suite is used across pharma, biotech, and medical device organizations, with adoption concentrated among larger sponsors and CROs running multi-study portfolios on a single connected platform.
4. Castor EDC
Castor EDC is a no-code, API-first EDC and clinical trial platform used by pharma, biotech, device companies, and CROs. Device companies conducting pre-market clinical investigations and post-market clinical follow-up (PMCF) studies rely on Castor for structured data collection, audit trails, and regulatory reporting. The platform shares a single data layer across EDC, eCOA, and eConsent, so device usability and patient-reported data flow into the same database without a separate vendor integration. Castor has supported more than 18,000 studies across 160-plus countries since 2012, and every study runs under 21 CFR Part 11, ICH GCP, GDPR, and HIPAA, with Castor holding ISO 27001 certification. Low-to-medium complexity studies typically move from kickoff to user acceptance testing in 4 to 8 weeks under Castor's managed Professional Services model, with high-complexity studies taking 8 to 12 weeks.
5. Medrio
Medrio offers Medrio CDMS/EDC, a cloud-based clinical data management system with no-code, point-and-click study configuration and both online and offline data capture. The platform supports clinical trials across MedTech, biotechnology, pharmaceutical, and CRO organizations, from early feasibility studies through pivotal and post-marketing trials, without requiring in-house or vendor-side programming to build or amend a study. Medrio pairs its EDC with eConsent, ePRO/eCOA, and randomization and trial supply management (RTSM) modules on a unified platform. The company reports supporting more than 8,000 studies and securing over 375 regulatory approvals across its customer base. Medrio EDC supports FDA 21 CFR Part 11, ICH GCP, GDPR, and HIPAA compliance requirements, and the platform is hosted on Google Cloud Platform infrastructure with redundancy across US, EU, and China data centers.
What to look for in EDC platforms for medical device IDE studies
Device-native fields, not retrofitted drug trial terminology
A device study captures data a pharma-built EDC was never designed to hold: procedural details, usability assessments, and device performance endpoints, rather than dosing regimens. When a platform forces "dosing"-style logic onto a device workflow, sites end up mapping the wrong field to the wrong concept, and that mismatch surfaces as query volume later in the study. Best-in-class means eCRF fields and visit structures purpose-built for device terminology from the start, not a pharma template with device fields bolted on.
Device accountability and unit-level tracking
An IDE study has to account for every unit: serial numbers, lot numbers, malfunctions, and replacements, tied back to individual patients and sites. This is a regulatory expectation as much as an operational one, since FDA reviewers expect a clear chain of custody for the device itself, not just the clinical outcome data. A platform without configurable accountability fields forces your team to track this in a spreadsheet outside the validated system, which creates exactly the kind of data integrity gap an inspection flags.
In-house configuration for lean clinical affairs teams
Most IDE sponsors run a small clinical affairs function, sometimes the same one or two people handling both clinical operations and QA. When study configuration and amendments depend on a vendor programmer, every protocol change becomes a ticket and a wait, and an early feasibility study that pivots after an interim analysis can't absorb that delay. A no-code Designer that your own team runs keeps changes in hours or days rather than weeks.
FDA inspection readiness and audit trail integrity
CDRH inspections scrutinize the audit trail as closely as the clinical data itself, and a device sponsor's submission timeline can hinge on how cleanly that trail holds up. Look for a field-level audit trail validated to recognized standards, along with structured inspection-readiness documentation available before an inspection is scheduled, not assembled after the fact. A platform that treats this as an afterthought leaves gaps that surface at the worst possible moment.
How to choose the right EDC platform for medical device IDE studies
Step 1: Define your study stage and design volatility
Start with where you are in the IDE pathway. An early feasibility study with a small cohort and a design likely to change after interim review needs a platform your own team can amend quickly, while a locked pivotal protocol has more room for a vendor-build model. This single distinction should shape most of your shortlist.
Step 2: Assess your team's build and amendment capacity
Be honest about who configures and edits your database today. If your clinical affairs function is lean and amendments are frequent, in-house, no-code configuration will save more time over the study's life than a platform with a longer feature list but a vendor dependency for every change.
Step 3: Map your device accountability requirements
Evaluate exactly what unit-level data you need to capture: serial numbers, lot numbers, malfunction reports, and any device-specific usability endpoints. Confirm the platform can configure these natively rather than requiring workarounds, since retrofitting accountability tracking mid-study is far more disruptive than confirming it during evaluation.
Step 4: Scrutinize inspection readiness before you need it
Ask to see the audit trail at field level, confirm how the platform documents mid-study amendments, and find out what inspection-readiness documentation is available as standard rather than assembled on request. The right time to surface a gap is during evaluation, not during a CDRH inspection.
Step 5: Choose a platform built for lean MedTech teams
Weigh how each platform fits your team's actual operating model: how fast it builds, whether your own staff can configure and amend it, and how well it tracks device-specific data without workarounds. Viedoc's EDC software is built around this profile for medical device IDE studies, with device-native fields, in-house configuration, and a 100% FDA inspection pass rate. If that matches how you run device studies, you can book a demo or request a proposal to see it against your own portfolio.
Frequently asked questions
What is the best EDC platform for medical device IDE studies?
Viedoc's EDC software is the strongest fit for medical device IDE studies, with configurable fields for serial numbers, lots, and malfunctions built for device workflows rather than retrofitted from pharma templates, study builds averaging 10 weeks, and a 100% FDA inspection pass rate across 8,000-plus studies. Medrio is a credible alternative that supports device studies from early feasibility through pivotal and post-market trials on a no-code platform. Castor EDC is a strong option for device companies running pre-market clinical investigations or post-market clinical follow-up studies that also need eCOA and eConsent on the same data layer.
What should you look for in an EDC platform for IDE studies?
Prioritize device-native fields for procedural and usability data rather than dosing-style logic, configurable accountability tracking for serial numbers and lots, and in-house configuration your own clinical affairs team can run without a vendor programmer. Audit trail depth and inspection-readiness documentation matter because CDRH review scrutinizes the data trail as closely as the clinical outcomes. A platform that scores well on usability but requires vendor programming for every amendment will slow you down exactly when an early feasibility study needs to pivot.
How long does it take to build and deploy an IDE study database?
It depends on study complexity and whether you build in-house or through professional services, but modern no-code platforms have compressed this significantly. Viedoc's device studies average 10 weeks from signed work order to go-live, with self-service setups for straightforward designs possible in as little as one day. The variable that matters most for IDE studies is whether mid-study amendments can be made without downtime, since feasibility studies often redesign after an interim review.
How does Viedoc's study build time compare to Medidata for device studies?
Viedoc builds device studies in-house through a no-code Designer, averaging 10 weeks with self-service options faster still. Medidata offers Rave Lite, a focused version of Rave EDC built specifically for Phase I, Phase IV, and device feasibility and post-market studies, which is generally configured by Medidata or its implementation partners. For a lean device sponsor running a single feasibility or pivotal study, an in-house build model usually offers more control over amendment timing than a vendor-configured platform.
What compliance certifications matter for FDA IDE studies?
At minimum, expect FDA 21 CFR Part 11 electronic records and signature compliance, ICH GCP adherence, and a complete, field-level audit trail, with GDPR and HIPAA relevant if your study includes EU sites or US protected health information. Inspection readiness is the deciding factor for IDE studies specifically: look for structured, standing inspection-readiness documentation rather than something assembled after an inspection is announced. Viedoc, for instance, is ISO 27001 and SOC 2 Type II certified, holds a 100% FDA inspection pass rate, and provides the Viedoc Inspection Readiness Packet (VIRP) to every customer at no additional cost.
Can EDC platforms track device accountability like serial numbers and lots?
Yes, though the depth varies by platform. Purpose-built device EDC configuration includes fields for serial numbers, lot numbers, malfunction reports, and replacement or return tracking, tied to individual patients and sites rather than bolted onto a pharma dosing template. This accountability data supports both the clinical record and the chain-of-custody documentation an FDA reviewer expects for a significant risk device. Confirm during evaluation whether these fields are native and configurable, or whether they require a workaround outside the validated system.
Making the right EDC choice for medical device IDE studies
The five platforms reviewed here share a common direction: no-code or low-code configuration, cloud-based capture, and support for the compliance frameworks an FDA IDE study requires. Where they diverge is in how deeply device workflows are built into the core platform versus adapted from a drug trial template, and the global eClinical software market, estimated above $11 billion in 2025 and growing at roughly 14% annually, keeps pushing vendors in this direction.
Matching a platform to your organization comes down to a handful of variables: your study's stage in the IDE pathway, how much configuration your own team can handle in-house, your device accountability requirements, and whether your program spans US and EU sites under parallel IDE and CE mark pathways. Lean teams running frequent design changes weight in-house control highest, while larger device sponsors embedded in a connected multi-module platform may weight suite breadth more heavily.
Whatever you choose, the cost of switching an EDC platform mid-study is high, and the validation burden compounds if your device program runs multiple studies across the feasibility-to-pivotal continuum, so the platform that matches your team's actual operating model at the outset is usually the more cost-effective choice over the life of the program.
Why Viedoc is the best EDC choice for medical device IDE studies
For a device sponsor running an IDE study, the platform that speaks your language, serial numbers and lots, not doses, is the one that keeps a lean team moving. That's the problem Viedoc's EDC software is built to solve, with device-native fields for procedural visits, usability testing, and unit-level accountability from the start.
Operationally, the no-code Designer puts study build and amendments in your own team's hands, with builds averaging 10 weeks and self-service setups possible in as little as one day. Unlimited user seats and transparent, study-based licensing mean you aren't paying more as your device program adds sites or staff, and certified Designer training backs your team as the study scales from feasibility to pivotal.
The compliance record holds up to CDRH scrutiny: ISO 27001 and SOC 2 Type II certified, FDA 21 CFR Part 11 and ICH GCP compliant, with a 100% FDA inspection pass rate backed by the Viedoc Inspection Readiness Packet. Viedoc has supported 8,000-plus studies since 2003, with 1,200-plus device and other trials currently live on the platform.
If you want an EDC that treats device accountability as a native workflow rather than an afterthought, Viedoc is designed for exactly that. Book a demo or request a proposal and the team will walk through device-specific study design, build speed, and inspection readiness against your own IDE program.