A notified body doesn't care that your electronic data capture (EDC) platform was built around dosing schedules and pharmacovigilance forms designed for a pill, not a device. When your post-market clinical follow-up (PMCF) study under the European Union Medical Device Regulation (EU MDR) needs device accountability tracking, years-long follow-up windows, and an audit trail that survives technical documentation review, a pharma-retrofitted EDC becomes the bottleneck. Viedoc's EDC software is built for this kind of long-horizon, device-specific data collection, with a no-code Designer your team runs independently and a compliance stack spanning GDPR, ISO 27001, GAMP 5, and FDA 21 CFR Part 11. This comparison evaluates five EDC platforms for EU MDR compliance across study design flexibility, audit trail depth, data residency, and total cost of ownership.
You're not running a single pivotal trial and moving on. Under EU MDR, PMCF is a continuous obligation that runs for the life of the device, feeding your Clinical Evaluation Report year after year, often across a portfolio of CE-marked products at different follow-up stages. Your platform needs to handle that duration without a fresh vendor contract or validation cycle every time a new obligation lands on your desk.
Enterprise EDC platforms built for large multi-country pharma trials carry timelines and overhead calibrated for far larger budgets than most device manufacturers run. Commodity or academic-grade tools rarely carry the validated audit trail and GDPR-compliant hosting a notified body expects. EU MDR clinical investigations and PMCF programs need a platform that's audit-ready by default, flexible enough to model device-specific data, and priced for a device manufacturer's study volume.
Best EDC solutions for EU MDR compliance: quick comparison
| Platform | Product / module | Overview |
|---|---|---|
| Viedoc | EDC Software | No-code EDC for device-specific study design, GDPR, ISO 27001, GAMP 5, and 21 CFR Part 11 compliant, across 7,500-plus studies. |
| Medidata | Rave EDC | Enterprise EDC supporting 23,000-plus MedTech sites, with device and diagnostic expertise built into the Medidata Platform. |
| Veeva | Vault EDC | EDC module within the Veeva Vault Clinical Suite, unifying data entry, imaging, and coding for device trials. |
| Castor | Castor EDC | No-code EDC with a dedicated PMCF offering for EU MDR post-market data collection and audit-ready exports. |
| Medrio | Medrio EDC | Cloud-based EDC suite with no-code builds and data ingestion from wearables and lab systems. |
These five EDC platforms represent the most evaluated options for medical device companies pursuing EU MDR compliance, reviewed across device-specific study design, audit trail depth, data residency, and total cost of ownership.
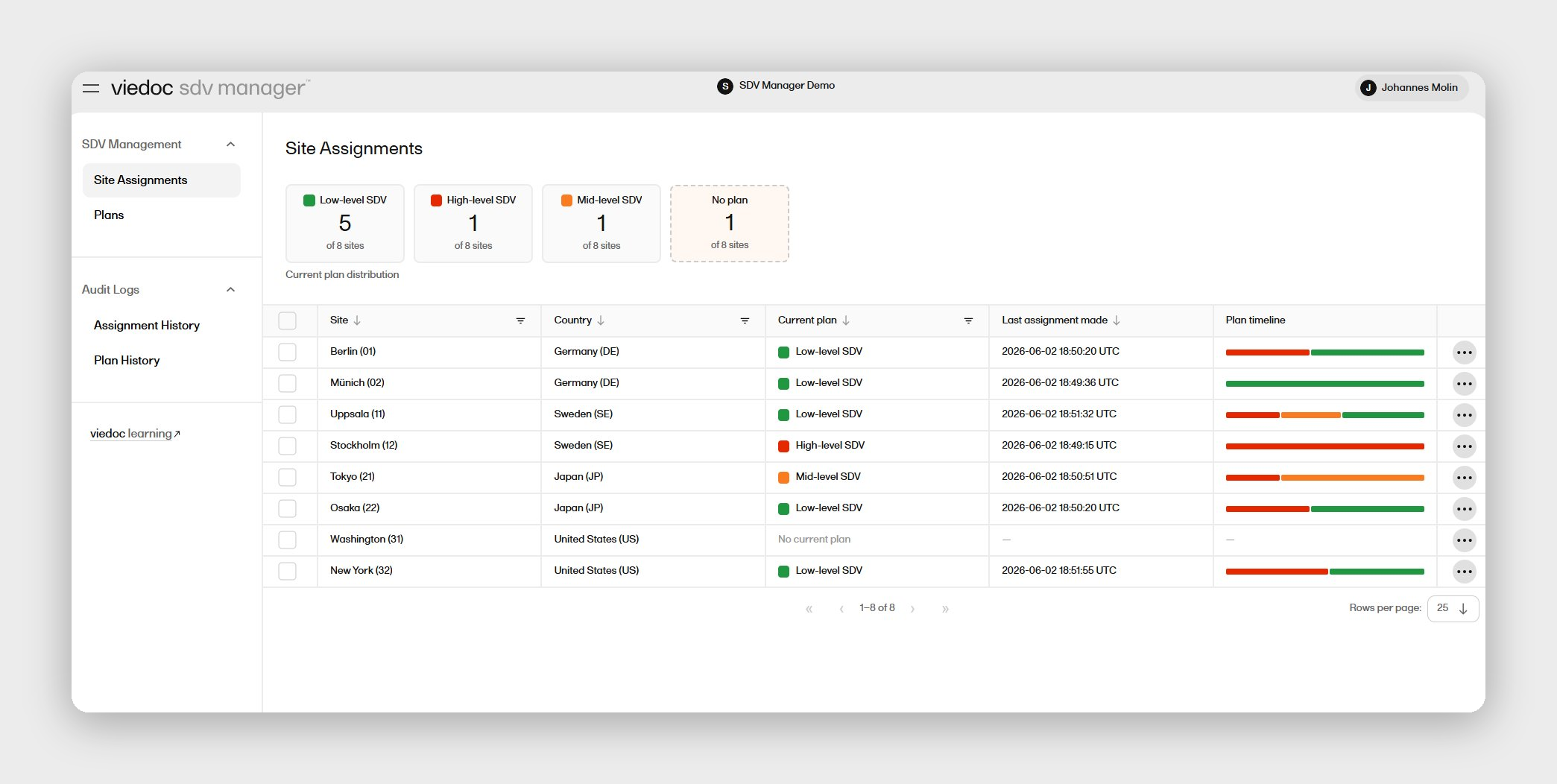
1. Viedoc
Viedoc's EDC software runs on a no-code Designer that lets your team configure device-specific forms, procedural visits, and accountability fields without pharma terminology in the way. The platform has powered more than 7,500 global studies across 75-plus countries, with study builds typically completed in 8 to 12 weeks through professional services, averaging 10 weeks. That build speed matters when a new PMCF obligation lands mid-portfolio and you need a validated database running in weeks, not the roughly 90-day standard tied to legacy enterprise EDC platforms.
For EU-based device manufacturers, Viedoc's GDPR-compliant hosting and European regulatory track record reduce the friction of running multi-country PMCF studies on a single validated system. The Viedoc Inspection Readiness Packet (VIRP) gives your QA team structured audit-readiness documentation at no additional cost, so a notified body review isn't a scramble to assemble evidence after the fact. Unlimited user seats and study-based licensing mean a lean team isn't paying a per-seat premium as PMCF follow-up scales.
Viedoc is certified to ISO 27001 and SOC 2 Type II, with GAMP 5-aligned validation, FDA 21 CFR Part 11 compliant electronic signatures, and ICH GCP and GDPR compliance built in. The platform supports 40-plus languages across 75-plus countries, and Viedoc's 24/7 customer success team provides direct escalation paths rather than a ticket-only queue.
"Audit trail generation in PDF format is a good feature." Dr. Vijay K., Assistant General Manager, made that assessment as a verified Viedoc user on G2.
-
Study scale: 8,500-plus studies run on Viedoc across 75-plus countries
-
Build speed: Study builds typically completed in 8 to 12 weeks through professional services, averaging 10 weeks
-
Compliance: FDA 21 CFR Part 11, ICH GCP, GDPR, ISO 27001, SOC 2, and GAMP 5
-
Inspection readiness: Viedoc Inspection Readiness Packet (VIRP) available to all customers at no additional cost
2. Medidata
Medidata offers Rave EDC, the electronic data capture product at the center of the Medidata Platform, supporting more than 23,000 MedTech sites worldwide through medical device and diagnostic-specific expertise built into the platform. Rave EDC integrates with Medidata's eConsent, eCOA, and imaging applications within a single validated environment, giving device sponsors a unified view across data sources rather than separate point solutions. The company also offers Rave Lite, a simplified configuration aimed at Phase I, Phase IV, feasibility, and post-market studies for teams that don't need a full enterprise build. Medidata's professional services and accreditation programs support CROs and device sponsors running studies through the platform across the full trial lifecycle, from early feasibility work through post-market follow-up, and the company reports serving pharmaceutical, biotechnology, and medical device customers globally.
3. Veeva
Veeva offers Vault EDC as part of the Veeva Vault Clinical Suite, alongside CTMS and eTMF applications on a single cloud platform. Vault EDC unifies data entry, image management, and automated coding within Veeva's MedTech Clinical Data offering, which is built specifically for device and diagnostic studies rather than adapted from a pharma-first product. The platform supports drag-and-drop study design and connects to the Veeva RIM Platform, giving device manufacturers a route from clinical data collection through to regulatory submission and global compliance tracking within the same vendor ecosystem. Vault EDC is designed for organizations already operating within, or planning to adopt, the broader Veeva ecosystem across clinical, quality, and regulatory functions, and the company reports adoption among a substantial share of the largest medical device and diagnostics manufacturers.
4. Castor EDC
Castor EDC is a cloud-native, no-code electronic data capture platform with a dedicated PMCF offering built specifically for post-market clinical follow-up under EU MDR. The platform pairs EDC with electronic patient-reported outcomes and audit-ready data exports designed for notified body review, and Castor states its Catalyst AI tool can reduce PMCF chart review costs by up to 80%. Castor is ISO 27001 certified, FDA 21 CFR Part 11 compliant, and supports GDPR and EU Annex 11 requirements, alongside ICH GCP alignment for interventional studies. The platform's no-code eCRF builder allows device manufacturers to configure and amend studies without a programmer or implementation consultant, and Castor positions the platform for sponsor-led device programs, prospective registries, and academic-adjacent investigator-initiated trials that need a validated system without enterprise-scale overhead.
5. Medrio
Medrio provides a cloud-based electronic data capture and eClinical suite spanning EDC, eCOA/ePRO, eConsent, and RTSM within a single database. The platform supports no-code, drag-and-drop study builds and automated ingestion of data from wearables, sensors, and external lab systems directly into the study record, reducing manual reconciliation for study teams. Medrio is used across pharmaceutical, biotech, medical device, diagnostics, and animal health study programs, and the company states its platform supports FDA 21 CFR Part 11 compliance for electronic records and signatures. Medrio's published materials on EU MDR-specific requirements are more limited than some other platforms in this comparison, with its public content weighted more toward US regulatory context, Phase I studies, and lower-complexity device programs rather than multi-country EU post-market follow-up.
What to look for in EDC solutions for EU MDR compliance
Long-horizon PMCF and post-market study design
A PMCF study isn't a fixed-duration interventional trial. It runs for the life of the device, often years, and needs to accommodate protocol amendments as new safety signals or usage patterns emerge without a full rebuild each time. Best-in-class platforms let your team amend forms and visit schedules directly, in days rather than through a vendor change order that can take weeks to turn around.
A platform that can't handle registry-style, open-ended follow-up will force your team into workarounds, like closing and reopening studies, that create exactly the kind of data fragmentation a notified body flags during review.
Device-specific data capture and accountability tracking
Device studies track serial numbers, lot numbers, malfunctions, and replacement events, not dosing and drug accountability. A platform retrofitted from pharma-first logic often forces your team to repurpose drug-accountability fields for device tracking, which creates confusing forms and downstream query volume. Best-in-class EDC platforms include native device accountability fields and configurable adverse-event and vigilance reporting workflows built for device terminology from the outset.
Audit trail depth and validation documentation for notified body review
Every action in the system, from a data entry to a signature to a query resolution, needs a complete, exportable audit trail your QA team can hand to a notified body without weeks of preparation. Best-in-class platforms maintain continuous, structured validation documentation as a standing artifact rather than something assembled ahead of an inspection. A platform that treats audit readiness as an afterthought turns every review into a scramble, and a weak audit trail can delay Clinical Evaluation Report sign-off.
EU data residency and GDPR-compliant hosting
Multi-country PMCF studies recruiting across the European Economic Area need a platform with GDPR-compliant hosting and a clear data processing agreement your legal and QA teams can review quickly. Best-in-class platforms publish their hosting infrastructure and certifications openly, rather than requiring a lengthy security questionnaire cycle before you can even evaluate fit.
How to choose the right EDC solution for EU MDR compliance
Step 1: Define your study type across the MDR lifecycle
Start by identifying whether you're running an initial clinical investigation to support CE marking, an ongoing PMCF study, or a broader post-market surveillance registry. Each has a different duration and amendment profile, and the platform that suits a fixed-term investigation may not suit a registry that needs to run for a decade.
Step 2: Assess your validation and audit-trail requirements
Ask your QA and regulatory affairs team what a notified body will expect to see in your technical documentation, and confirm the platform's validation package matches that bar before you commit. A platform that requires you to build your own validation evidence from scratch adds months to your timeline that a validated, audit-ready system avoids.
Step 3: Evaluate device-workflow fit against your protocol
Walk through your actual case report form requirements, including device accountability, usability endpoints, and adverse event terminology, rather than a generic demo. A platform built around pharma dosing logic will show its limits the moment you try to model a real device workflow.
Step 4: Scrutinize data residency and total cost of ownership
Confirm where your data is hosted, how the pricing model scales as PMCF follow-up extends across more years and more sites, and whether unlimited user seats are included as your team grows. Hidden per-seat or per-amendment fees erode your budget precisely as your post-market obligations compound.
Step 5: Choose a platform your team can run independently
Once you've mapped your study type, validation bar, and workflow requirements, choose the platform your own clinical team can configure and maintain without depending on vendor programmers for every amendment. Viedoc's EDC software is built for exactly this kind of self-sufficiency across the EU MDR lifecycle, from CE-marking investigations through years of PMCF follow-up. Book a demo or request a proposal to see it against your own device portfolio.
Frequently asked questions
What is the best EDC platform for EU MDR compliance?
Viedoc's EDC software is the best choice for medical device companies managing EU MDR clinical investigations and PMCF studies, combining a no-code Designer your team configures independently with a validated compliance stack covering GDPR, ISO 27001, GAMP 5, and FDA 21 CFR Part 11 across 7,500-plus studies. Castor EDC is a credible alternative with a dedicated PMCF offering built specifically around EU MDR post-market data collection. Medidata is the enterprise benchmark for larger, multi-market device programs, though its build timelines and professional-services model are calibrated for bigger organizations than most device manufacturers running a PMCF program.
What should I look for when choosing an EDC platform for EU MDR clinical investigations?
Look for device-specific form configuration, a continuous and exportable audit trail, GDPR-compliant hosting, and a validation package your QA team can hand to a notified body without extra preparation. Pricing transparency matters too, since PMCF obligations often run for years and unpredictable per-seat or per-amendment fees compound over that duration.
How long does it take to build and deploy a study database for a PMCF or post-market registry?
It depends on the platform and delivery model. Self-service teams on modern no-code platforms can have a database live in as little as a day, while professional-services builds on validated enterprise platforms typically run 8 to 12 weeks. Legacy enterprise platforms calibrated for large pharma programs can run closer to 90 days for a full build.
What compliance certifications matter most for EU MDR clinical investigations and PMCF studies?
At minimum, look for FDA 21 CFR Part 11 for electronic records and signatures, ICH GCP, GDPR for EU data handling, ISO 27001 for information security, and GAMP 5 for computer system validation. GAMP 5 alignment matters particularly for device studies, since it directly addresses the validation approach notified bodies expect from computerized systems supporting technical documentation.
Is there such a thing as an "EU MDR certified" EDC platform?
Not exactly. EU MDR certification applies to the medical device itself, granted through a notified body's conformity assessment, not to the software used to collect clinical data. What you're evaluating in an EDC platform is whether its validation, audit trail, and data-handling practices produce evidence a notified body will accept as part of your technical documentation, not a certification badge on the software itself.
Which EDC platforms support post-market clinical follow-up (PMCF) studies?
Viedoc, Castor EDC, Medidata, and Veeva all support post-market and observational study designs alongside standard interventional trials, though the depth of PMCF-specific tooling varies. Castor EDC publishes a dedicated PMCF offering built around EU MDR requirements, while Viedoc supports PMCF and post-market workflows through the same flexible, no-code EDC platform used for pre-market clinical investigations.
Making the right EDC choice for EU MDR compliance
The platforms in this comparison share a common baseline of GDPR compliance and validated audit trails, but they differ sharply in how much of that validation burden falls on your team versus the vendor, and in how well their data models fit device workflows. The global eClinical software market is valued at more than $11 billion and continues to grow at a double-digit annual rate, with EU MDR compliance pressure a significant driver of demand among device manufacturers.
The right fit depends on your organization's size, the number of CE-marked products in your post-market portfolio, and how much validation work you want in-house versus outsourced. Larger manufacturers running concurrent PMCF programs across markets tend to weight suite depth and enterprise support more heavily, while lean device teams typically weight self-service configuration and predictable pricing.
Switching EDC platforms mid-portfolio carries real validation cost, so getting this choice right before your first PMCF obligation lands avoids a disruptive re-validation cycle later.
Why Viedoc is the best EDC choice for EU MDR compliance
When a PMCF obligation spans years and multiple CE-marked products, you need an EDC software platform built for that duration from day one, not one retrofitted from pharma logic. Viedoc's no-code Designer lets your clinical team configure device-specific forms, accountability fields, and amendments independently, backed by 7,500-plus completed studies across 75-plus countries and 99.99% platform uptime.
Unlimited user seats and transparent, study-based licensing mean your costs stay predictable as PMCF follow-up scales across more sites and more years, without per-seat fees penalizing a growing lean team. The Viedoc Inspection Readiness Packet (VIRP) keeps your QA team's validation evidence current and exportable, so a notified body review doesn't turn into weeks of scrambling for documentation. Certified to ISO 27001 and SOC 2 Type II, with GAMP 5-aligned validation, GDPR compliance, and FDA 21 CFR Part 11 electronic signatures, Viedoc gives your team a validated foundation across the full EU MDR lifecycle.
If you're managing clinical investigations and PMCF studies across a growing device portfolio and want a platform your own team can run without vendor dependency, book a demo or request a proposal and we'll walk through your study design, validation requirements, and total cost of ownership together.