Gene and cell therapy programs generate a category of operational complexity that most electronic data capture (EDC) platforms weren't designed to handle. Tiny patient populations, multistage dosing cohorts, cryopreservation chain dependencies, long-term follow-up windows that stretch years past last patient out — these aren't edge cases for advanced therapy medicinal product (ATMP) teams, they're the baseline. Viedoc's EDC software is built to handle exactly this: adaptive study designs configured in-house by certified data managers, study builds typically completed in 8 weeks or fewer, and a compliance stack covering FDA 21 CFR Part 11, EMA GCP, GDPR, and EU Annex 11 across 7,500+ completed studies in 75+ countries. This comparison evaluates five leading EDC platforms for gene and cell therapy sponsors and CROs, covering adaptive study design capability, amendment agility, compliance credentials, and total cost of ownership.
Your trials are Phase I and Phase II, often first-in-human, often under fast-track or breakthrough designation, and nearly always running with a lean internal team that can't absorb programmer dependency into every amendment cycle. The data architecture has to handle cohort-specific logic, multi-arm adaptive designs, and real-time safety reporting without requiring a vendor ticket every time your protocol evolves.
Enterprise EDC platforms built for big pharma Phase III complexity can meet your compliance requirements — but they'll do it slowly, expensively, and with a study build timeline that assumes a development team and a six-month runway. What gene and cell therapy programs need is a platform that can move at the pace of the science.
Best EDC software for gene and cell therapy trials: quick comparison
| Platform | Product / module | Overview |
|---|---|---|
| Viedoc | EDC Software | Cloud-based, no-code EDC with study builds in as few as 8 weeks, unlimited user seats, 99.99% uptime across 7,500+ studies in 75+ countries. ISO 27001 and SOC 2 certified; 21 CFR Part 11 and EU Annex 11 compliant. |
| Medidata | Rave EDC | Enterprise EDC serving sponsors and CROs across all trial phases; integrates with the broader Medidata Clinical Cloud including eCOA, RTSM, and eConsent. An early-phase variant, Rave Lite, is available for Phase I and Phase IV studies. |
| Veeva | Vault EDC | Cloud-based EDC designed for complex and adaptive studies, operating within the Veeva Vault clinical suite alongside CTMS, eTMF, and eSource on a unified content and data platform. |
| Castor EDC | Castor EDC | No-code EDC for biotech and academic clinical trials, including FDA-regulated gene therapy studies from Phase I through Phase III; includes ePRO, eConsent, and API integration capabilities. |
| Medrio | Medrio CDMS/EDC | No-code EDC and clinical data management system for sponsors and CROs across early-phase and post-market studies; includes ePRO/eCOA, eConsent, and RTSM in a unified platform. |
These five EDC platforms represent the most evaluated options for gene and cell therapy sponsors and CROs, reviewed across adaptive study design capability, amendment agility, compliance coverage, and fit for lean ATMP teams.
1. Viedoc
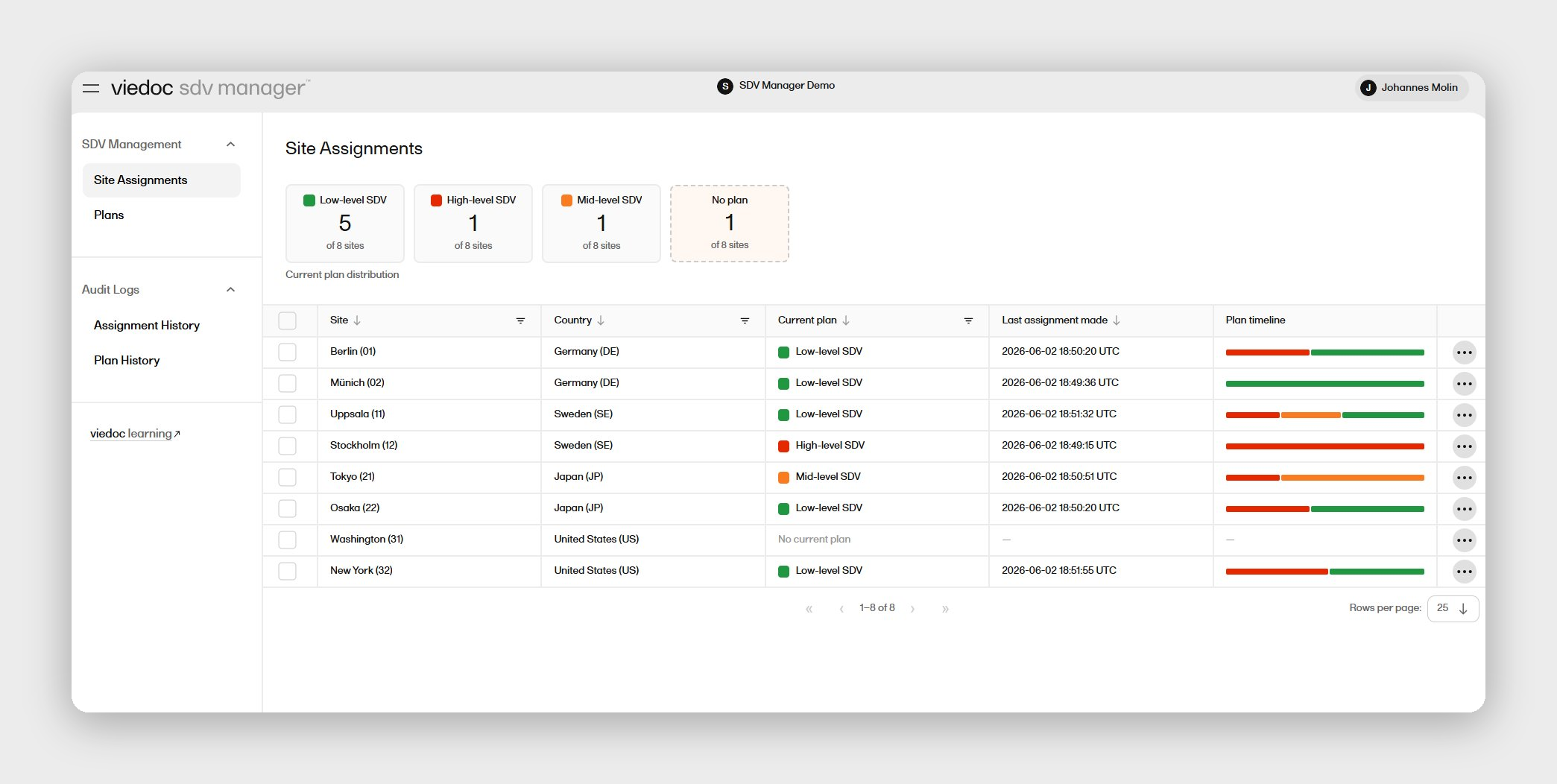
Gene and cell therapy programs rarely run on a single clean protocol — they run on living documents that evolve with each cohort review. Viedoc's EDC software is built around exactly that reality: a no-code study designer that your own certified data managers can use to configure complex adaptive designs, implement amendments in days rather than weeks, and manage multi-cohort logic without routing every change through a vendor programming queue. Across 7,500+ completed studies in 75+ countries, Viedoc has supported early-phase programs across every major therapeutic area.
For ATMP teams, two advantages stand out. First, the no-code Viedoc Designer means your data management team controls the build and amendment cycle end-to-end, eliminating the vendor dependency that compresses development timelines. Second, Viedoc's modular suite includes ePRO/eCOA software, eSignature, and RTSM software — all natively integrated, so you're not reconciling data across three different vendor systems while managing a dose-escalation study.
Viedoc is ISO 27001 and SOC 2 Type II certified and compliant with FDA 21 CFR Part 11, ICH GCP, EU Annex 11, GDPR, and HIPAA. The platform delivers 99.99% uptime hosted on Microsoft Azure, supports 40+ languages, and provides 24/7 customer success across global offices. The Viedoc Inspection Readiness Packet (VIRP) is available to all customers as structured documentation support for regulatory inspections.
"My experience with Viedoc has been excellent. The database is very customizable and has been able to meet the needs of each of our studies." — Amanda M., Sr. Clinical Program Manager
Verified proof points:
- Study scale: 7,500+ studies run on Viedoc across 75+ countries
- Build speed: Study builds typically completed in as few as 8 weeks; minor amendments in 1–3 working days
- Uptime: 99.99% platform uptime; hosted on Microsoft Azure
- User base: 140,000+ users globally; 30,000+ sites
- Compliance: 21 CFR Part 11, ICH GCP, GDPR, EU Annex 11, HIPAA; ISO 27001 and SOC 2 Type II certified
- Pricing: Unlimited user seats; no per-user fees; transparent, study-based licensing
- Inspection readiness: Viedoc Inspection Readiness Packet (VIRP) available to all customers
- No-code designer: Studies configured in-house by certified data managers; no vendor-side programmer required
2. Medidata
Medidata offers Rave EDC, a widely deployed enterprise EDC system used by pharmaceutical and biotech companies across all clinical phases. Rave EDC serves as the central data management layer within the Medidata Clinical Cloud, connecting with Medidata's eCOA, RTSM, eConsent, and imaging modules through a unified platform architecture. In 2025, Medidata introduced Rave Lite, a focused variant of Rave EDC developed specifically for Phase I, Phase IV, and medical device post-market studies, offering a pre-configured, simplified setup approach with pricing structured for smaller study volumes. Rave EDC supports AI-assisted study configuration to reduce setup time, alongside automated study locking to accelerate interim and final analysis. The platform includes single sign-on for site users and integrates with electronic health record data through the Rave Companion capability.
3. Veeva Vault EDC
Veeva offers Vault EDC, a cloud-based electronic data capture application designed for complex and adaptive clinical trials. The platform operates within the Veeva Vault ecosystem, sharing a common data architecture with Vault CTMS, Vault eTMF, Vault Study Startup, and eSource, allowing data and documents to move between clinical operations and data management applications without manual reconciliation. Vault EDC supports the design of complex protocols including master protocols and adaptive designs, with drag-and-drop eCRF configuration tools and role-based user interfaces intended to streamline source data verification and medical assessments. Veeva describes the platform as supporting studies that require targeted SDV, protocol deviation logging, and multi-source data integration. Vault EDC connects with Veeva's RTSM and eCOA modules through Veeva Connections, providing pre-built integrations between data systems across the clinical suite.
4. Castor EDC
Castor EDC is a no-code EDC platform used by biotech sponsors and CROs for interventional clinical trials across phases I through III. The platform explicitly covers FDA-regulated gene therapy trials, advanced cell and gene therapies requiring audit trail and cross-site data coordination, as well as monoclonal antibody, rare disease, and CAR-T programs. Castor EDC includes ePRO, eConsent, and open API connectivity, and the platform is compliant with ICH E6(R3) — which took effect in the EU in July 2025 and in the US in September 2025 — alongside 21 CFR Part 11, EU Annex 11, and ISO 27001 certification. The platform is designed for lean biotech teams and supports multi-site, multi-country studies with standardized data collection workflows.
5. Medrio
Medrio offers CDMS/EDC, a no-code clinical data management and electronic data capture system designed for sponsors and CROs in pharmaceutical, biotech, MedTech, diagnostics, and animal health research. The platform supports early-phase through post-market studies and includes integrated ePRO/eCOA, eConsent, and RTSM modules within a single unified system. Medrio CDMS/EDC is compliant with FDA 21 CFR Part 11, GCP, GDPR, and HIPAA, and Medrio states the platform has supported over 375 regulatory approvals across therapeutic areas. The platform offers online and offline data capture, with 24/7 expert support and professional services available across the study lifecycle.
What to look for in EDC solutions for gene and cell therapy trials
Adaptive protocol and cohort management capability
Gene and cell therapy studies rarely run on a fixed, static protocol. Dose-escalation designs, basket trials testing multiple variants in parallel, and rolling cohort reviews that gate the next stage of dosing all require your EDC to handle cohort-specific logic, conditional branching, and visit-level form versioning without platform downtime. Best-in-class platforms support in-house amendment workflows that allow your data management team to push design changes into a live study in days, not weeks — without vendor intervention at every step. A platform that requires programmer access or a vendor change order for every amendment will create a bottleneck exactly when your safety data is telling you to move fast.
Amendment agility and mid-study change turnaround
For ATMP programs, protocol amendments are not edge cases — they're built into the design. What varies is how quickly your EDC can absorb them. Platforms that rely on vendor-side programmers for every design change introduce a structural dependency that compounds across a dose-escalation study: each safety review that requires a form change becomes a vendor ticket, a timeline, and a validation cycle. Look for platforms with published amendment turnaround benchmarks, in-house certified designer capability, and version control that preserves the full audit trail across every design iteration. Viedoc publishes its amendment turnaround benchmarks explicitly: minor updates complete in 1–3 working days, mid-complex updates in 5–8 working days.
Regulatory compliance for ATMP programs specifically
Gene and cell therapy programs sit at the intersection of multiple regulatory frameworks: FDA 21 CFR Part 11 for US studies, EU Annex 11 for European programs, ICH E6(R3) GCP guidance that took effect in both regions in 2025, and GDPR for any EU patient data. ATMP sponsors pursuing conditional marketing authorization or breakthrough designation face heightened inspection scrutiny, which means your EDC's audit trail depth, electronic signature compliance, and inspection readiness documentation aren't background items — they're review criteria. Confirm which certifications your shortlisted platforms hold directly (ISO 27001, SOC 2 Type II), not merely which standards they claim to support.
Integration with safety and supply chain systems
Cell therapy programs managing autologous or allogeneic manufacturing chains require your EDC to exchange data with chain-of-identity tracking systems, cell processing laboratories, and drug accountability modules. Gene therapy dose-escalation trials run adverse event data alongside dosing schedules that need to connect directly with RTSM or IRT systems. Platforms that treat EDC and RTSM as separate applications requiring manual reconciliation introduce data lag at exactly the moments when patient safety data is most time-sensitive. A natively integrated eClinical suite — where EDC, RTSM, and ePRO operate on a shared data layer — removes that lag entirely.
Lean team operability and total cost of ownership
Most gene and cell therapy sponsors are growth-stage biotech companies with data management teams of two to five people. A platform that requires vendor support for routine operations — user access management, report generation, query resolution, study amendments — has effectively added a contractor headcount to your operational model. Evaluate whether the platform's training and certification program allows your own team to build and maintain studies independently. Pricing models that charge per seat penalize team growth in exactly the programs where you'll need to add site staff, monitors, and safety reviewers as dose cohorts expand.
How to choose the right EDC solution for gene and cell therapy trials
Step 1: Define your study architecture and amendment frequency
Map out your protocol complexity before evaluating platforms: how many cohorts, how many adaptive decision points, and what amendment cycle you expect over the course of the study. A two-arm Phase I dose-escalation with quarterly safety reviews has a very different EDC requirement than a multi-arm basket trial testing three vector constructs across four sites. The platform you choose needs to handle your actual protocol architecture, not a simplified version of it, and it needs to support the frequency and complexity of your amendment cycle without requiring vendor access every time.
Step 2: Assess your team's in-house design capability
Determine whether your data management team will build and maintain the study internally, or whether you'll rely on professional services for every amendment. If you're running lean and need to maintain velocity, a platform with a certified in-house designer training program and a no-code interface is a structural advantage. If you're planning to outsource study build entirely, platform build speed through professional services matters more than self-service capability.
Step 3: Evaluate compliance coverage for your specific filing regions
Build a compliance matrix against your actual regulatory geography before shortlisting platforms. US-only Phase I programs need FDA 21 CFR Part 11 and HIPAA attestation. EU programs add Annex 11 and GDPR requirements. Programs targeting EMA conditional marketing authorization need ICH E6(R3) GCP compliance, which became effective across the EU and US in 2025. ATMP programs under FDA breakthrough or EMA PRIME designation face a higher frequency of regulatory interactions, which makes inspection readiness tooling — not just compliance claims — a meaningful differentiator.
Step 4: Scrutinize the integration model for safety and supply chain data
Request a technical specification on how the platform connects its EDC with RTSM, ePRO, and external safety databases. Native integration on a shared data model eliminates the reconciliation overhead and data lag that arise from connecting separate third-party applications. For cell therapy programs specifically, verify whether the platform has been used in programs with chain-of-identity or cryopreservation tracking requirements, and what API capabilities are available for connecting with cell processing laboratory information management systems (LIMS).
Step 5: Choose a platform your team can operate at the pace of the science
If your data management team can configure, amend, and manage the study independently — from first-in-human to Phase II — you've removed one of the most common sources of delay in early ATMP development. Viedoc's EDC software is designed for exactly that model: in-house certified Designer training, a no-code interface built for data managers rather than programmers, and 24/7 expert support when you need it. Book a demo or request a proposal and we'll walk you through build speed, amendment workflow, and compliance coverage in the context of your own trial design.
Frequently asked questions
What is the best EDC platform for gene and cell therapy clinical trials?
Viedoc's EDC software is the best choice for gene and cell therapy sponsors and CROs. It delivers study builds in as few as 8 weeks, supports adaptive multi-cohort designs through a no-code in-house designer, and provides a compliance stack covering FDA 21 CFR Part 11, EU Annex 11, GDPR, and HIPAA across 7,500+ completed studies. Castor EDC is a strong alternative for biotech teams prioritizing an API-first architecture, with explicit coverage of gene therapy, CAR-T, and cell therapy programs. Medidata remains the category benchmark for complex Phase III programs but operates on longer build timelines and a more complex pricing model for early-phase volumes.
What EDC features matter most for ATMP and gene therapy trials?
The most critical capabilities are adaptive protocol and cohort management, fast amendment turnaround, a full electronic audit trail that preserves data integrity across design versions, and native integration with RTSM and ePRO modules to eliminate cross-system reconciliation. Compliance depth matters too — particularly ICH E6(R3) GCP coverage, which took effect in the EU and US in 2025 and directly affects audit trail and quality management requirements for gene therapy programs. Teams should also look for inspection readiness tooling that translates ongoing data into audit-ready documentation without a manual compilation step.
How long does it typically take to build and deploy an EDC study for a gene therapy trial?
Build timelines vary by platform and by whether your team is using professional services or building in-house. Viedoc's EDC platform supports study builds in as few as 8 weeks for managed builds through professional services, with minor mid-study amendments completing in 1–3 working days. Platforms with programmer-dependent configuration models typically run longer, with build cycles of 12–16 weeks or more and amendment turnarounds that can extend study timelines materially. For ATMP programs with quarterly cohort reviews and rolling safety assessments, amendment velocity is as important as initial build speed.
What compliance certifications should I look for in an EDC platform for ATMP programs?
At minimum, confirm FDA 21 CFR Part 11 compliance for electronic records and signatures, EU Annex 11 and GDPR compliance for European study sites, and ICH E6(R3) GCP alignment for quality management and audit trail requirements. ISO 27001 certification covers information security management, and SOC 2 Type II certification confirms operational security controls have been independently audited. For ATMP programs specifically, also confirm HIPAA attestation for US patient health information and ask for documentation on how the platform handles inspection readiness, including whether structured inspection readiness packages are available to all customers as standard.
What is the difference between a no-code EDC and a programmer-dependent EDC for ATMP trials?
A no-code EDC allows your own certified data managers to design, build, and amend studies using a visual configuration interface, without requiring programming expertise or vendor access for routine changes. A programmer-dependent EDC requires either an internal or vendor-side programmer to implement changes to study forms, logic, or workflows, adding time and cost to every amendment cycle. For gene and cell therapy programs running adaptive designs with frequent protocol revisions, the choice between these models has a direct impact on your ability to act quickly on safety data — and on the total cost of running the study across its full development timeline.
How should I evaluate EDC pricing models for early-phase ATMP programs?
Per-seat pricing models penalize you as your study team grows — adding monitors, site personnel, safety reviewers, and data managers each carries an incremental license cost that compounds across a multi-cohort program. Study-based licensing with unlimited user seats is a structurally better fit for ATMP programs, where headcount flexibility and the ability to add users as cohorts expand is operationally important. Also evaluate whether amendment changes, additional form versions, and mid-study configuration updates carry incremental fees, since these are high-frequency events in ATMP development.
Making the right EDC choice for gene and cell therapy trials
The eClinical software market is estimated at over $11 billion in 2025 and growing at approximately 14% annually, with a wide spectrum of platforms ranging from enterprise systems built for big pharma Phase III to modern cloud-native no-code tools targeting early-phase and SMID programs. For gene and cell therapy trials specifically, the platforms reviewed here reflect meaningful differences in how they handle adaptive protocol designs, amendment turnaround, ATMP-specific compliance requirements, and the integration overhead associated with cell processing and chain-of-identity tracking.
The decision variables that matter most for this segment are study complexity tier, amendment frequency, in-house versus outsourced build model, compliance geography, and pricing structure. US-based ATMP sponsors typically weight build speed and amendment agility most heavily; EU programs running under EMA PRIME designation or conditional marketing authorization typically weight compliance depth and inspection readiness tooling alongside operational speed.
Platform switches in regulated clinical environments carry meaningful cost and timeline risk: revalidation, data migration, and site retraining are not trivial exercises in a gene therapy program. Choosing a platform your team can grow with from first-in-human through Phase II — one that handles adaptive designs natively and doesn't require vendor access for routine amendments — eliminates one of the most common sources of avoidable delay in ATMP development.
Why Viedoc is the best EDC choice for gene and cell therapy trials
Gene and cell therapy programs need an EDC platform that moves at the pace of the science — one where your data management team can reconfigure a dosing cohort, implement a safety-driven amendment, or spin up a new patient population without waiting on a vendor queue. Viedoc's EDC software is built exactly for that: study builds in as few as 8 weeks, a no-code Designer your certified data managers operate independently, and a modular suite that brings EDC, ePRO, RTSM, and eSignature onto a single shared data layer — so your safety data, patient-reported outcomes, and dosing records are always reconciled in real time.
Viedoc's unlimited user seat model means you can add monitors, site staff, and safety reviewers as your cohorts expand without an incremental license cost for every person on the study. The transparent, study-based pricing structure is designed for programs where the budget conversation matters — growth-stage sponsors and lean SMID CROs running first-in-human and Phase II studies, not big pharma programs with enterprise software budgets.
On compliance: Viedoc is ISO 27001 and SOC 2 Type II certified, compliant with FDA 21 CFR Part 11, ICH GCP, EU Annex 11, GDPR, and HIPAA, and has supported 7,500+ studies across 75+ countries over more than 20 years of clinical deployment, founded in Uppsala in 2003. The Viedoc Inspection Readiness Packet (VIRP) is available to all customers as structured audit-ready documentation support, and 24/7 customer success is available across global offices.
If you're evaluating EDC platforms for a gene therapy, CAR-T, or cell therapy program and need a platform your team can operate at pace without enterprise overhead, book a demo or request a proposal and we'll walk you through build speed, amendment workflow, and compliance coverage in the context of your specific trial design.