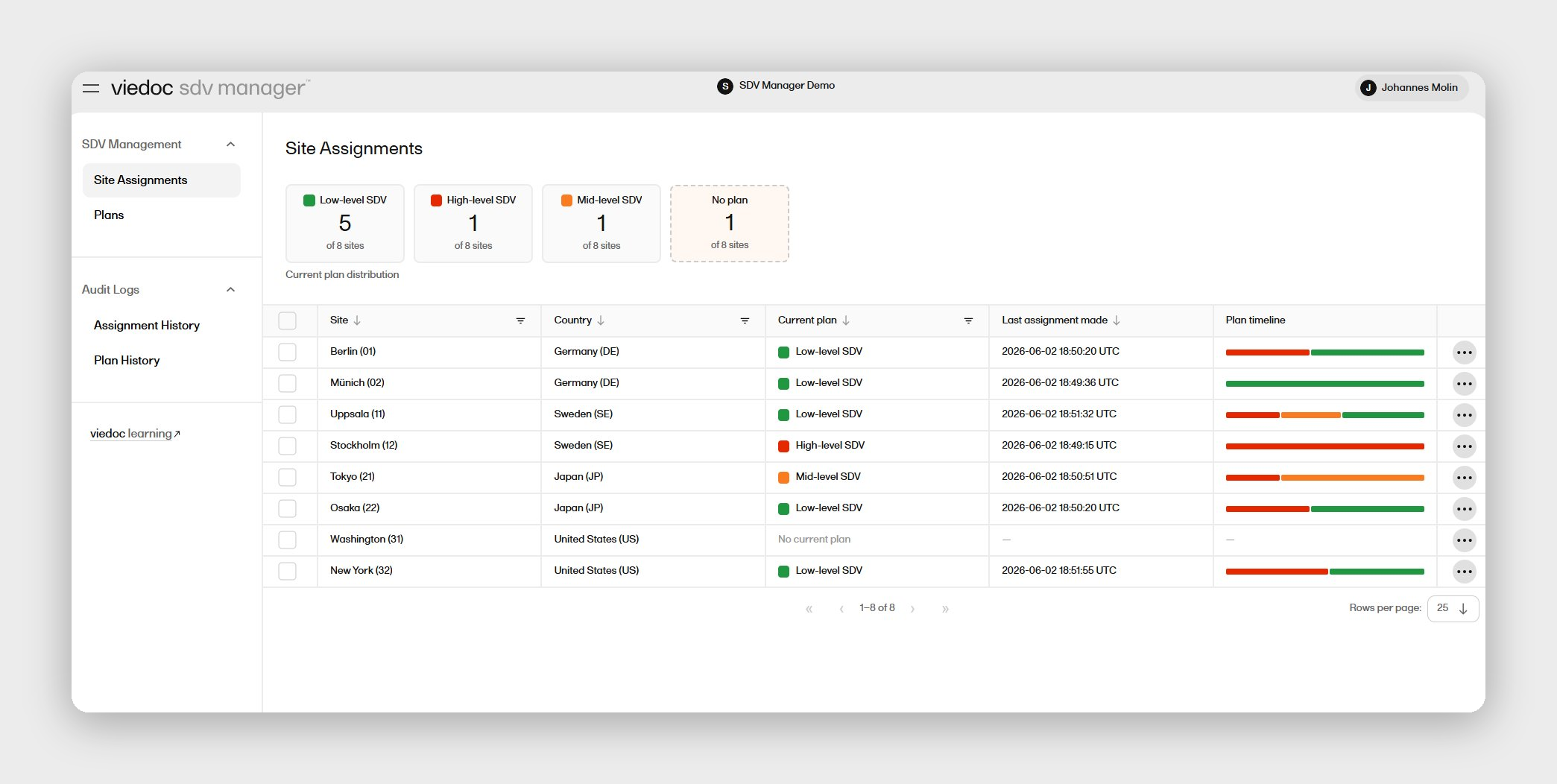
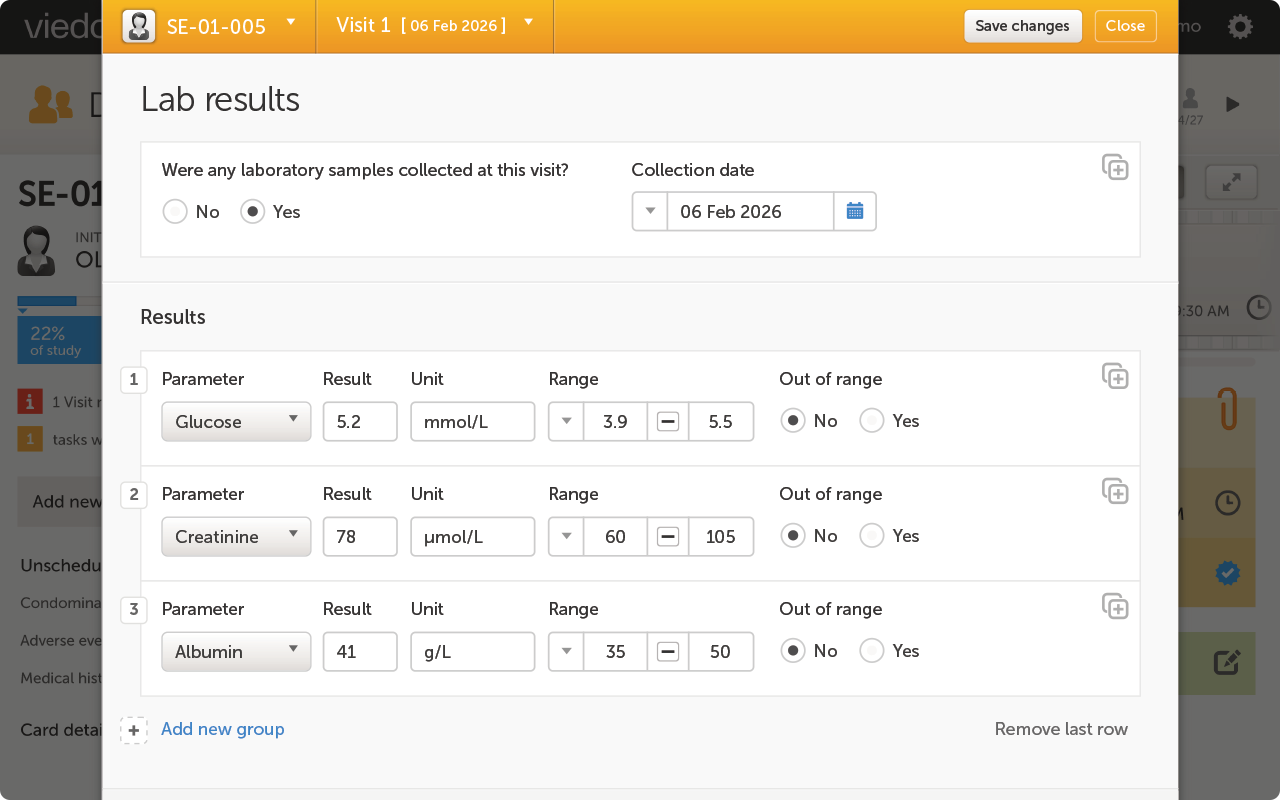
When you're running a rare disease trial, your electronic data capture (EDC) platform carries a specific burden that standard commercial trials don't. Small patient populations mean every enrolled participant is irreplaceable. Protocol amendments — and there are often many — can't wait 90 days for a vendor programmer to turn them around. And because your data will face FDA or EMA scrutiny with limited precedent, your audit trail, inspection readiness, and compliance documentation have to be airtight from day one. Viedoc's EDC software is purpose-built for exactly this operating environment: study builds typically completed in 2–4 weeks, no-code amendments your data management team can execute in-house, and a compliance stack covering 21 CFR Part 11, GDPR, EU Annex 11, and HIPAA across 75+ countries. This comparison evaluates six leading EDC platforms for rare disease clinical trials across amendment velocity, study design flexibility, patient engagement capability, and inspection readiness.
Rare disease programs move on tighter timelines than their large-population counterparts. You're often managing rolling Phase 1/2 designs, adaptive protocols, and global multi-site recruitment from a relatively lean team. The platform you choose needs to flex with your study — not create bottlenecks every time your protocol evolves.
Generic enterprise EDC solutions are architected for large, stable Phase III studies with heavyweight programming teams and 12-month build cycles. For rare disease teams, that architecture is misaligned from the start. What you need is fast study configuration, seamless amendment handling, multi-language patient engagement, and a compliance posture that holds up under regulatory scrutiny — without building a vendor dependency into every protocol change.
Best EDC solutions for rare disease trials: quick comparison
| Platform | Product / module | Overview |
|---|---|---|
| Viedoc | EDC Software | Cloud-native EDC with no-code study designer, 40+ languages, unlimited user seats, and study builds typically completed in 2–4 weeks across 7,000+ studies in 75+ countries. |
| Medidata | Rave EDC | Enterprise cloud EDC platform used extensively in large pharma; offers full feature depth including SDV, log-lines, and batch signing; standard build timelines of up to 90 days. |
| Veeva | Vault EDC | Cloud-based EDC within the Vault Clinical suite; strong integration with eTMF, CTMS, and RIM modules; suited to sponsors already operating within the Veeva ecosystem. |
| Castor EDC | Castor EDC | No-code, API-first cloud EDC with self-service study design; supports Phases I–IV; used by CROs and smaller biotech/pharma; strong in real-world evidence and registry studies. |
| Medrio | Medrio EDC | User-friendly cloud EDC targeting Phase I and Phase II trials and CROs; supports offline data capture and is available in 100+ countries; includes ePRO, eConsent, and RTSM modules. |
| Oracle | Oracle Clinical One | Enterprise EDC platform with open architecture supporting integration across CTMS, supply chain, and safety systems; historically strong in large North American pharma studies. |
These six EDC platforms represent the most commonly evaluated options for rare disease clinical trial teams, reviewed across amendment speed, study design flexibility, multilingual patient engagement, regulatory compliance, and inspection readiness.
1. Viedoc
For rare disease trial teams, the compounding cost of platform friction is felt acutely: a single delayed amendment can stall data collection for patients who cannot simply be replaced by broader enrollment. Viedoc's EDC software is designed around self-sufficiency — a no-code Designer that your certified data managers configure and amend in-house, without routing every change through a vendor programming queue. Study builds typically complete in 2–4 weeks, and amendments are handled in days, not months.
For rare disease programs that frequently run rolling Phase 1/2 designs and adaptive protocols, Viedoc's modular suite adds particular depth. Viedoc's ePRO solution supports 40+ languages with BYOD capability, automated reminders, and wearable device integration — directly relevant when patient populations are geographically dispersed and every compliance data point matters. Viedoc's randomization software supports both static and dynamic randomization within the same study, with no separate login required. When a Clinfidence-managed Phase 1 trial for Citryll navigated 17 mid-study protocol amendments, Viedoc absorbed each change without disrupting data collection — and the Phase 1 eCRF template was reused to reduce Phase 2a setup time by up to 30%.
Viedoc is ISO 27001 and SOC 2 certified, and compliant with 21 CFR Part 11, GDPR, EU Annex 11, HIPAA, ICH GCP, and EMA guidelines. The Viedoc Inspection Readiness Packet (VIRP) is available to all customers as structured audit-readiness documentation, giving lean QA teams a structured head start on computer system validation (CSV) and regulatory submission preparation. 24/7 customer support is available across global offices, with direct escalation pathways rather than a ticket-only model.
"My experience with Viedoc has been excellent. The database is very customizable and has been able to meet the needs of each of our studies. Customer Support has been excellent with quick turnarounds and response time." — Amanda M., Sr. Clinical Program Manager
Verified proof points:
- Study scale: 7,000+ studies run on Viedoc across 75+ countries
- User base: 140,000+ users globally; 30,000+ sites
- Build speed: Study builds typically completed in 2–4 weeks
- Uptime: 99.99% platform uptime; hosted on Microsoft Azure
- Language support: Available in 40+ languages
- Pricing: Unlimited user seats; no per-user fees; transparent, study-based licensing
- Compliance: 21 CFR Part 11, GDPR, Annex 11, HIPAA; ISO 27001 and SOC 2 certified
- Inspection readiness: Viedoc Inspection Readiness Packet (VIRP) available to all customers
- Support: 24/7 support across global offices
- No-code designer: Studies configured in-house by certified data managers; no vendor-side programmer required
2. Medidata
Medidata offers Rave EDC, a cloud-based electronic data capture platform with broad adoption across large pharmaceutical and biotech organizations. Rave EDC provides full feature depth including source data verification (SDV), log-lines, batch signing, and protocol deviation management, which are standard requirements in complex Phase III trials. The platform supports integration with Medidata's broader clinical cloud suite, including safety, imaging, and patient engagement modules. Study build timelines on Rave EDC typically run up to 90 days and require vendor-side programmers for configuration and amendments, which can create resourcing pressure for rare disease teams managing frequent protocol changes on tight timelines. Medidata's pricing structure is enterprise-oriented and per-seat, which adds cost variability as study teams and CRO partners scale.
3. Veeva
Veeva offers Vault EDC as part of its Vault Clinical suite, a cloud-based platform designed to integrate EDC with eTMF, CTMS, and regulatory information management (RIM) in a single environment. Vault EDC is built on a modern, zero-downtime cloud architecture and carries compliance credentials including 21 CFR Part 11, GDPR, and ISO certifications. The suite integration model is particularly relevant for sponsors already using Vault eTMF or CTMS, where data flows seamlessly across the platform. Vault EDC is primarily positioned at mid-to-large pharma and biotech sponsors; the premium pricing model reflects full-suite licensing, which can represent overhead for rare disease teams seeking standalone EDC capability without the broader Vault investment.
4. Castor EDC
Castor EDC offers a no-code, API-first cloud EDC platform with self-service study design and a drag-and-drop form builder. The platform supports Phases I through IV and is used by CROs and smaller biotech and pharma organizations, with particular strength in real-world evidence, registry studies, and medical device trials. Castor provides ePRO, eConsent, and direct data capture capabilities alongside its core EDC module, and maintains REST API connectivity for integrations with electronic health record (EHR) and eConsent systems. The platform's self-service model reduces vendor dependency for study builds, though its track record in large-scale, heavily regulated commercial rare disease trials is less established than platforms with deeper enterprise deployment histories.
5. Medrio
Medrio offers a cloud-based EDC platform designed for Phase I and Phase II trials, with a focus on usability for CROs and smaller biotech and pharma organizations. The platform includes ePRO, eConsent, direct data capture, and RTSM modules alongside core EDC functionality, and supports offline data capture for sites with connectivity constraints. Medrio is available in 100+ countries and has expanded its service model to include clinical trial project management support and study build services. The platform's primary strength is in lower-complexity, early-phase studies; its feature set for large, multi-regional rare disease studies with complex randomization and adaptive design requirements is less documented in publicly available materials.
6. Oracle Clinical One
Oracle Health offers Clinical One, a cloud-based EDC platform with an open architecture supporting integration across CTMS, supply chain, and safety systems via a digital gateway and microservices design. Clinical One targets large pharmaceutical and biotech organizations, primarily in North America, and supports all study phases and therapeutic areas. The platform provides structured integration pathways with Oracle's broader health and life sciences portfolio. Oracle's product strategy has shifted toward the wider consumer health market in recent years, which has raised questions in the industry about the long-term investment trajectory for Clinical One specifically, particularly for mid-market and rare disease buyers outside North America.
What to look for in EDC software for rare disease clinical trials
Amendment velocity and in-house configurability
In rare disease development, protocol amendments are not the exception — they're built into the development arc. You're often running first-in-human or first-in-class studies where the protocol evolves in response to emerging safety and pharmacodynamic data. What matters is how quickly your EDC can absorb those changes, and whether your own team can make them without routing every modification through a vendor programming queue. Best-in-class platforms let certified data managers build and amend studies in-house; the benchmark for initial study build is 2–4 weeks, with amendments processed in days. The cost of a vendor-dependent amendment cycle, measured in data collection delays across a population you cannot easily expand, is operationally significant in a way it isn't for large Phase III programs.
Small-N study design flexibility
Rare disease trials require electronic case report form (eCRF) designs that don't follow standard templates. You may be capturing novel biomarker endpoints, disease-specific scoring systems, complex multi-stage dose escalation workflows, or assessments that have never been formalized across a validated EDC platform before. Your platform needs a no-code or low-code designer capable of handling branching logic, custom calculations, and stage-dependent visit schedules — and it needs to be flexible enough to absorb design changes mid-study without triggering a full revalidation cycle. Platforms that require vendor programmers for non-standard form configurations introduce a latency risk that's disproportionate for rare disease programs.
Multilingual ePRO and patient engagement
Rare disease populations are almost always globally distributed. With patient pools that may number in the dozens rather than hundreds, your trial frequently spans multiple countries and languages, and every enrolled participant's continued engagement is critical to data completeness. An integrated ePRO solution with broad language support, BYOD compatibility, automated reminders, and real-time compliance monitoring is not a nice-to-have in this context — it's fundamental to data integrity. Look for platforms supporting 40+ languages natively, with patient interfaces that don't require specialist hardware provisioning and work on participants' own devices.
Inspection readiness and audit trail depth
Rare disease submissions often face additional scrutiny because of the limited clinical evidence base available at the time of approval. Your EDC's audit trail, change control documentation, and computer system validation (CSV) support have to hold up under FDA, EMA, and PMDA review — often without the precedent of previous submissions in the same indication. Platforms that provide pre-built validation documentation, structured inspection readiness packages, and audit trails that are complete, exportable, and searchable reduce the burden on lean QA teams and accelerate your submission timeline. Verify that the platform's VIRP or equivalent documentation covers the EMA Guideline on Computerised Systems and FDA Q&A Guidance on Electronic Systems as a minimum.
Pricing model alignment with rare disease program economics
Rare disease programs are rarely single-study programs, but they're also rarely large-enrollment programs. A pricing model that charges per enrolled subject, per active user, or per amendment penalizes the exact characteristics of a rare disease trial portfolio: small enrollment, frequent protocol changes, and lean study teams that expand and contract across studies. Transparent, study-based licensing with unlimited user seats means your cost structure doesn't compound unpredictably as your team grows, your protocol evolves, or you add CRO partners to your preferred vendor network.
How to choose the right EDC software for rare disease clinical trials
Step 1: Define your amendment frequency and team configuration
Map how frequently your program typically generates protocol amendments and whether your data management team is resourced to handle those changes in-house. If you expect iterative amendments across a multi-stage Phase 1/2 design, a platform requiring vendor-side programmer involvement for every change will create compounding delays. Define whether self-service configuration is a firm requirement before evaluating platforms.
Step 2: Assess your patient engagement architecture
Confirm whether your study requires integrated ePRO, patient diaries, or wearable data capture alongside core EDC. For rare disease programs with geographically dispersed patient populations, the gap between a basic EDC and a fully integrated ePRO/EDC suite is significant — both in data completeness and in regulatory defensibility. If ePRO is required, evaluate platforms where patient data flows directly into the EDC without manual reconciliation or a separate system.
Step 3: Evaluate your compliance and validation baseline
Confirm which regulatory frameworks apply to your program — FDA 21 CFR Part 11, EMA GCP, EU Annex 11, HIPAA, GDPR — and verify that each shortlisted platform carries the certifications to match. For rare disease submissions with limited precedent, pre-built validation documentation and inspection readiness tooling reduce your QA team's workload materially. Ask vendors specifically how they support IQ/OQ/PQ validation and what inspection readiness documentation is available to customers without additional cost.
Step 4: Scrutinize total cost of ownership across your program lifecycle
Request full pricing transparency across the study lifecycle, including amendment fees, user seat costs, module add-ons, and support tier pricing. Compare the cost of a self-service platform where your team builds and amends studies in-house against an enterprise platform where vendor services fees accumulate with every change. For rare disease programs running multiple studies concurrently across a small team, the difference in total cost of ownership over a three-to-five-year program can be substantial.
Step 5: Choose a platform that scales with your program without re-validation
Select a platform that can support your program from Phase 1 through Phase 2 and beyond without requiring full system re-validation at each transition. The ability to reuse eCRF templates across study phases — and to carry your compliance documentation forward — reduces timeline and cost at each phase transition. Viedoc's EDC software supports this model, with the no-code Designer enabling template reuse across studies and VIRP documentation that travels with the platform rather than requiring a rebuild at each new phase. Book a demo or request a proposal to discuss your specific program requirements.
Frequently asked questions
What is the best EDC software for rare disease clinical trials?
Viedoc's EDC software is the best choice for rare disease clinical trials, combining study builds typically completed in 2–4 weeks, no-code in-house amendment capability, 40+ language ePRO integration, and a full compliance stack covering 21 CFR Part 11, GDPR, EU Annex 11, and HIPAA across 7,000+ studies in 75+ countries. The transparent, study-based pricing model with unlimited user seats aligns well with rare disease program economics, where small patient populations and frequent protocol amendments make per-seat or per-amendment pricing structures costly. Medidata is the benchmark for large pharma Phase III programs with complex feature requirements including full SDV and batch signing, but its 90-day build timelines and programmer-dependent amendment model are structurally misaligned with rare disease development timelines. Castor EDC is a credible alternative for early-phase or registry-style rare disease studies with a self-service configuration model.
How long should it take to build and deploy an EDC study for a rare disease trial?
On a modern, no-code EDC platform, study builds for early-phase rare disease trials should be completable in 2–4 weeks from protocol receipt to go-live. That window assumes an in-house certified data manager using a self-service designer — not a vendor programming queue. Amendment turnaround on the same platform should be measurable in days, not weeks. Legacy enterprise platforms that require vendor-side programmers for initial builds and each subsequent amendment routinely operate on 60–90 day build timelines, which is incompatible with the iterative nature of most rare disease Phase 1 programs.
What eCRF design features matter most for rare disease studies?
Rare disease eCRF design requires branching logic for complex multi-stage dose escalation, custom scoring calculations for disease-specific endpoints, stage-dependent visit schedules, and the ability to add or modify assessments mid-study without triggering full system revalidation. Platforms with drag-and-drop no-code designers allow your data management team to build and validate these configurations in-house. The ability to reuse and template eCRF designs across study phases — carrying Phase 1 architecture forward into Phase 2 — reduces both build time and validation overhead at each transition.
What compliance certifications should a rare disease EDC platform carry?
For rare disease trials targeting FDA or EMA approval, the minimum compliance baseline is 21 CFR Part 11, ICH GCP, EU Annex 11 (for EU studies), GDPR (for EU patient data), and HIPAA attestation (for US patient health information). ISO 27001 certification for information security management and SOC 2 Type II certification for data handling controls are the standard independent verifications of platform security posture. For programs with Japanese sites or post-market surveillance components, PMDA ERES compliance and APPI (Japan) coverage are additionally required. Verify that the platform's inspection readiness documentation covers each applicable regulatory framework explicitly.
What is the Viedoc Inspection Readiness Packet (VIRP) and is it relevant for rare disease submissions?
The Viedoc Inspection Readiness Packet (VIRP) is a structured set of audit-readiness documentation available to all Viedoc customers, covering system validation, regulatory compliance evidence, and documentation aligned to FDA, EMA, and PMDA inspection expectations. For rare disease submissions — where regulatory reviewers may be working with limited prior precedent in the indication — having pre-structured, complete validation documentation reduces the QA team's workload and accelerates submission timelines. VIRP covers the EMA Guideline on Computerised Systems and Electronic Data in Clinical Trials, the FDA Q&A Guidance on Electronic Systems, and PMDA expectations as a baseline.
Can a single EDC platform support a rare disease program from Phase 1 through Phase 2 without re-validation?
Yes, provided the platform is built on a cloud-native architecture with version-controlled study design and a documented change-control audit trail. Platforms that allow eCRF template reuse across study phases, carry validation documentation forward, and support protocol amendments without full system revalidation reduce both the timeline and cost burden at each phase transition. This is a specific advantage for rare disease programs that move quickly between Phase 1 and Phase 2 and cannot afford the 6–12 month revalidation cycles that legacy on-premise systems typically require.
Making the right EDC choice for rare disease clinical trials
The EDC platforms reviewed here represent a range of architectures — from enterprise suite-first platforms with deep Phase III feature sets to no-code, cloud-native solutions optimized for fast study builds and self-service amendment management. What they have in common is a cloud-hosted delivery model, regulatory compliance capability, and support for the core data capture, query management, and audit trail functions that all regulated clinical trials require.
The decision framework for rare disease teams is shaped by program-specific variables: amendment frequency and in-house team capacity, geographic spread of patient populations and language requirements, the regulatory frameworks applicable to your target markets, and a pricing model that aligns with the economics of small-enrollment, multi-phase programs. US-based rare disease sponsors typically weight study build speed and self-service configurability most heavily; EU-based programs frequently lead with compliance documentation depth and EU Annex 11 credibility. Neither evaluation is wrong — they reflect different regulatory pressure points.
Rare disease programs also carry a switching cost that's higher than most. Migrating an active study mid-program, or rebuilding validated systems at a phase transition, consumes QA and data management resources that rare disease teams rarely have to spare. Choosing a platform that can serve the full program lifecycle — and carry its own validation documentation forward — is worth a more thorough evaluation upfront.
Why Viedoc is the best EDC choice for rare disease clinical trials
If you're building a rare disease program on a platform that requires a vendor programmer for every amendment, you're not just paying more — you're building a structural bottleneck into a development arc where timelines are already compressed. Viedoc's EDC software puts amendment control back in the hands of your data management team. Study builds typically complete in 2–4 weeks. Amendments happen in days. And the eCRF templates you build for Phase 1 can be carried forward into Phase 2, reducing setup time and revalidation overhead at each transition.
Viedoc's modular suite adds the depth rare disease programs need beyond core EDC: ePRO/eCOA with 40+ language support and wearable integration, randomization and RTSM with dynamic allocation capability, Televisits for geographically dispersed patient populations, and Medical Coding with MedDRA and WHODrug integration — all within a single validated environment. Unlimited user seats and transparent, study-based pricing mean your cost structure stays predictable as your program and team scale.
Viedoc is ISO 27001 and SOC 2 certified, and compliant with 21 CFR Part 11, GDPR, EU Annex 11, HIPAA, ICH GCP, and EMA guidelines — with VIRP documentation available to all customers for inspection-ready CSV support. Founded in 2003, Viedoc has supported 7,000+ studies across 75+ countries, with 99.99% platform uptime and 24/7 customer success support backed by direct escalation pathways.
If your rare disease program needs an EDC that moves at the pace of your science rather than your vendor's programming queue, book a demo or request a proposal. Our team will walk you through amendment speed, compliance credentials, and ePRO integration in the context of your specific study design.