When a device approval timeline slips because your electronic data capture (EDC) platform can't handle a non-standard visit schedule, or because every protocol amendment needs a vendor programmer, that's not a software problem — it's a platform fit problem. Viedoc's EDC software is purpose-built for the flexibility that device studies require, with a no-code Designer your clinical team configures independently, a native Japan post-market surveillance module unique among SMID-focused EDC vendors, and a compliance stack covering FDA 21 CFR Part 11, GDPR, HIPAA, ISO 27001, and GAMP 5 — all deployed across 7,000+ studies in 75+ countries. This comparison evaluates six leading EDC platforms for medical device and MedTech teams across study design flexibility, lean-team usability, Japan and APAC regulatory support, compliance credentials, and total cost of ownership.
Device studies are structurally different from pharmaceutical trials. You're managing usability assessments, diagnostic performance studies, post-market clinical follow-up (PMCF), and post-market surveillance (PMS) programs — often with a lean clinical team carrying all the operational weight. The EDC platform you choose will either work with those workflows or force you to work around them.
Most EDC platforms are built for pharma. Terminology is calibrated for dosing, not device accountability. Visit structures assume fixed interventional windows, not the long-horizon follow-up of a post-market registry. Platforms built for Medidata's 90-day build cycle don't compress well when a PMCF study needs to launch in weeks, not quarters. What device teams need is right-sized: fast to configure, adaptable by design, and compliant without enterprise overhead.
Best EDC solutions for medical device trials: quick comparison
| Platform | Product / module | Overview |
|---|---|---|
| Viedoc | EDC Software + PMS (Japan) | Cloud-native EDC with no-code study design, a native Japan post-market surveillance module, and full compliance with FDA 21 CFR Part 11, GDPR, HIPAA, ISO 27001, and GAMP 5. Used in 7,000+ studies across 75+ countries. |
| Medidata | Rave EDC | Enterprise EDC platform used in large-scale pharma and device trials, with validated compliance tooling and a broad partner ecosystem. |
| Medrio | Medrio EDC | Cloud-based EDC designed for Phase I and lower-complexity trials, with no-code configuration and offline data capture capability. |
| Castor EDC | Castor EDC | API-first, no-code EDC platform with self-service study configuration, used across pharma and some device research settings. |
| Veeva | Vault EDC | Cloud EDC integrated within the Veeva Vault suite, with connections to Veeva's CTMS, eTMF, and regulatory information management tools. |
| Oracle Health | Oracle InForm | Long-established enterprise EDC platform with deep regulatory compliance tooling, used across large pharma and CRO portfolios. |
These six eClinical platforms represent the most commonly evaluated options for medical device and MedTech teams, reviewed across study design flexibility, Japan and APAC regulatory support, lean-team configurability, compliance credentials, and total cost of ownership.
1. Viedoc
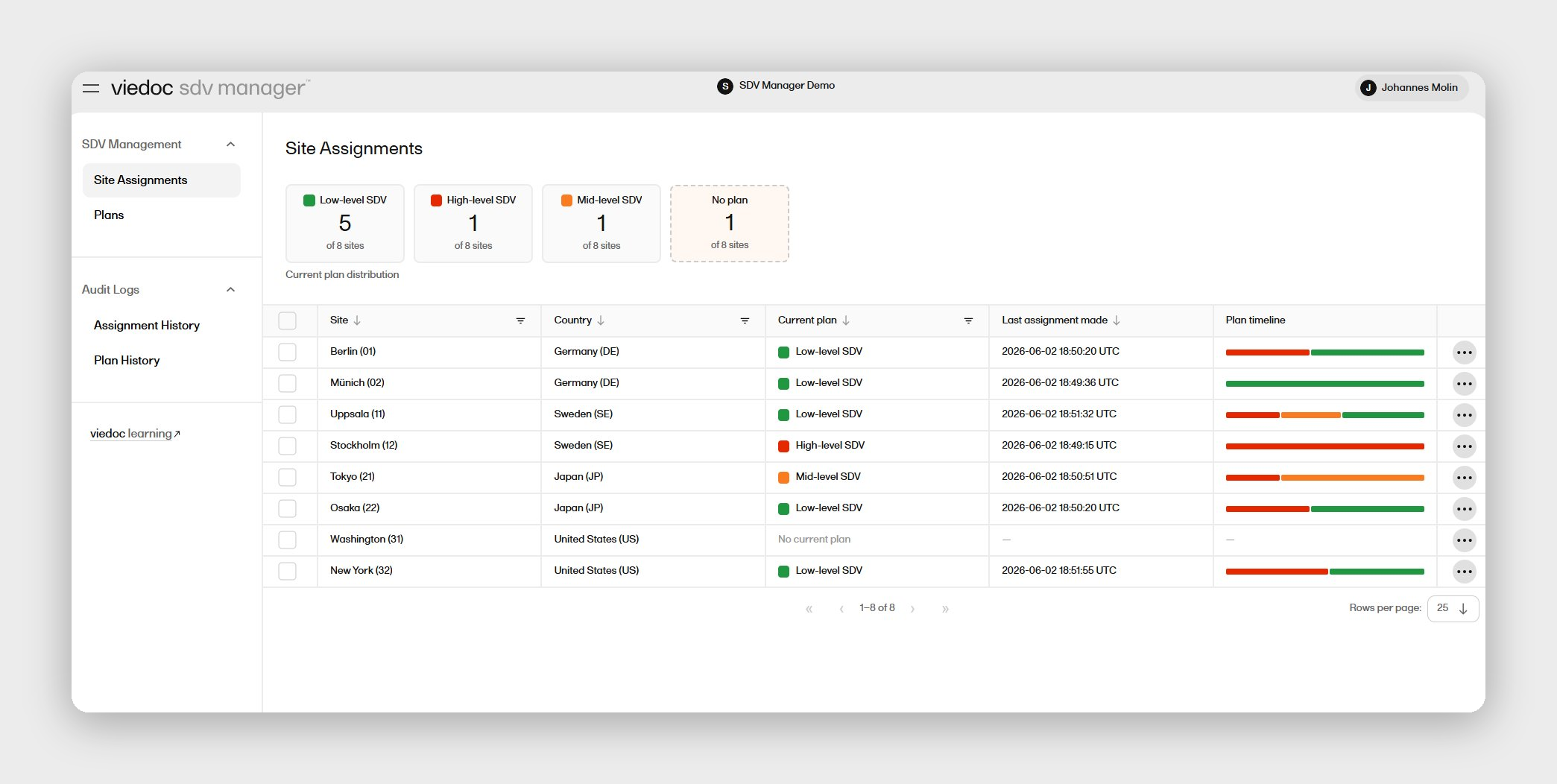
Viedoc's EDC software supports medical device studies across all phases and study types — including diagnostic performance studies, usability assessments, investigational device exemption (IDE) trials, CE mark studies, and post-market programs — with a no-code Designer that lets your clinical data management team configure and amend studies in-house, without a vendor programmer on the critical path. The platform has supported 7,000+ studies across 75+ countries, with 1.6 million trial participants and 30,000+ sites — including medical device studies run by MDCE, a global CRO managing neurosurgery, ophthalmology, and endocrinology device trials on Viedoc across nine European sites.
Two capabilities set Viedoc apart for this segment specifically. The native Japan post-market surveillance module — Viedoc PMS — handles booklet data collection, the Kaifu process, and PMDA ERES-compliant reporting within the standard Viedoc Designer environment, making it the only SMID-focused EDC platform with native Japan PMS support. Equally important for lean clinical teams: the unlimited user seat model means you're never pricing in headcount as your study grows, and the Viedoc Inspection Readiness Packet (VIRP) provides structured audit-readiness documentation to every customer.
Viedoc is certified ISO 27001 and SOC 2, compliant with FDA 21 CFR Part 11, ICH GCP, EU Annex 11, GDPR, HIPAA, GAMP 5, and APPI (Japan), and operates at 99.99% uptime on Microsoft Azure with 24/7 customer support across global offices. The platform is available in 40+ languages, supporting device teams running studies across APAC, Europe, and the US from a single system.
"My experience with Viedoc has been excellent. The database is very customizable and has been able to meet the needs of each of our studies." — Amanda M., Sr. Clinical Program Manager
Verified proof points:
- Study scale: 7,000+ studies across 75+ countries; 30,000+ sites
- Build speed: Study builds typically completed in 2–4 weeks; 10-week average for full professional services delivery
- Uptime: 99.99% platform uptime; hosted on Microsoft Azure
- Japan PMS: Native Viedoc PMS module for Japan post-market surveillance; PMDA ERES compliant; no additional designer training required
- Compliance: FDA 21 CFR Part 11, ICH GCP, GDPR, HIPAA, EU Annex 11, GAMP 5, ISO 27001, SOC 2, APPI (Japan)
- Pricing: Unlimited user seats; no per-user fees; transparent, study-based licensing
- Inspection readiness: VIRP available to all customers
- Support: 24/7 support across global offices
2. Medidata
Medidata offers Rave EDC, an enterprise electronic data capture platform with an established presence across large-scale pharmaceutical, biotechnology, and medical device trials. Rave EDC provides audit trail management, electronic signatures, and integration with Medidata's broader Clinical Cloud suite — including modules for safety, CTMS, and patient engagement. The platform supports FDA 21 CFR Part 11 and ICH GCP compliance and is used by large global CROs and pharmaceutical companies running complex multi-regional trials. Medidata's study build process typically involves vendor-side programmers and configuration timelines that can extend to 90 days for more complex builds, which reflects the depth of customization available but introduces a timeline consideration for device teams operating under compressed launch schedules. Medidata does not offer a native Japan post-market surveillance module.
3. Medrio
Medrio offers a cloud-based EDC platform designed for Phase I and lower-complexity clinical trials, with no-code study configuration and support for offline data capture. The platform covers EDC, ePRO, eConsent, and RTSM within a unified database, and has been used in device trials and early-phase sponsor studies, primarily in the US market. Medrio supports FDA 21 CFR Part 11 compliance and includes flexible data review workflows and custom reporting. The platform has limited global presence outside of North America, with a smaller footprint in Europe, Japan, and APAC compared to Viedoc. Medrio does not publish a dedicated Japan post-market surveillance module on its website, and the platform's documentation of complex study logic and large multi-regional study support is less comprehensive than enterprise incumbents.
4. Castor EDC
Castor EDC offers an API-first, no-code EDC platform with self-service study configuration and a free-tier option for academic and lower-complexity studies. The platform provides drag-and-drop form building, branching logic, and automated data validation, with support for ePRO and eConsent. Castor EDC is used in pharmaceutical and some device research contexts, primarily for studies of lower-to-moderate complexity. It holds ISO 27001 certification and supports GDPR compliance. Castor's suite depth is narrower than platforms with integrated RTSM, eTMF, or Japan PMS capability, and its track record in large multi-site regulated device trials is less documented than more established platforms.
5. Veeva Vault EDC
Veeva offers Vault EDC as part of the Veeva Vault cloud platform, which spans EDC, CTMS, eTMF, regulatory information management, and safety. Vault EDC is built for organizations already operating within the Veeva ecosystem and provides a connected data environment across the clinical trial lifecycle. The platform supports FDA 21 CFR Part 11 and ICH GCP compliance and is positioned for mid-to-large pharmaceutical and biotechnology sponsors. Vault EDC's value proposition is strongest for organizations adopting Veeva's full suite; standalone EDC buyers who don't need adjacent Vault modules may find the platform's commercial model less favorable. Veeva does not publish a dedicated Japan post-market surveillance module on its website.
6. Oracle InForm
Oracle Health offers Oracle InForm, a long-established enterprise EDC platform with validated compliance tooling, deep audit trail functionality, and integrations across Oracle's broader health and life sciences portfolio. InForm has been used in large pharma and CRO programs across multiple therapeutic areas and supports FDA 21 CFR Part 11 and ICH GCP compliance. The platform operates with a legacy architecture that requires substantial IT and programmer resource to configure and maintain, which increases deployment lead times and amendment overhead for lean device teams. Oracle InForm is positioned for large-scale enterprise programs and is less commonly evaluated for lower-complexity device studies or lean-team MedTech environments.
What to look for in EDC solutions for medical device and MedTech trials
Study design adaptability for non-pharma workflows
Device studies don't map cleanly onto pharma-centric eCRF templates. Your study might involve device accountability tracking with serial numbers and lot numbers, usability assessment endpoints, diagnostic performance measurements, or post-market observational follow-up over several years. A platform that forces you to retrofit pharma-standard CRF structures onto device-specific data types compounds your configuration work before you even reach go-live. Best-in-class EDC for device teams provides genuinely flexible form and workflow design — configurable visit structures, device-specific field types, and logic that supports the study you're actually running. If a demo doesn't let you demonstrate your own study's data model within the first session, that's a signal.
Lean-team configurability and vendor independence
Most MedTech clinical operations teams are small. A single Director of Clinical Affairs may own the EDC relationship, the regulatory submission timeline, and the budget. A platform that requires a vendor programmer for every amendment, or a multi-week change control process for minor CRF updates, doesn't scale to that operating model. Look specifically at how the platform handles mid-study changes: what's the turnaround for a minor CRF update? Can your data manager implement it without opening a support ticket? Viedoc's no-code Designer allows certified in-house data managers to configure and amend studies independently — minor updates typically take one to three working days without vendor involvement. That independence is a direct operational advantage for lean teams.
Japan and APAC regulatory coverage
If your device program includes a Japan approval pathway, PMDA expansion, or post-market surveillance obligation under Japan's Pharmaceutical and Medical Device Act (PMD Act), your EDC platform needs to handle Japan-specific PMS requirements natively — including booklet data collection formats, the Kaifu review process, and PMDA ERES-compliant reporting. Most pharma-centric EDC platforms don't support these workflows without significant custom development. Viedoc's dedicated Japan PMS module handles these requirements within the standard Designer environment, with no additional training needed for teams already certified in Viedoc. For device companies with any Japan footprint, this is a meaningful differentiator worth testing in a product demonstration.
Compliance documentation and inspection readiness tooling
The compliance stack for device trials spans FDA 21 CFR Part 11, ICH GCP, EU MDR and IVDR requirements, GDPR, HIPAA for US studies, GAMP 5 for computer system validation, and APPI for Japan. Each of these imposes audit trail, validation, and data integrity obligations that your EDC platform must satisfy — and that your QA team will verify before sign-off. Beyond the certifications themselves, look at what inspection-readiness infrastructure is included as standard. Pre-built validation documentation, structured computer system validation (CSV) support, and organized audit-readiness packages reduce your QA team's workload at inspection time. Viedoc's VIRP is structured exactly for this and is available to all customers at no additional charge.
Total cost of ownership for study-based and portfolio work
Per-user pricing models penalize you as your study team grows. Per-study models that charge setup fees for every amendment add unpredictable costs to programs that frequently change. For device teams running multiple studies simultaneously — including some at different stages of regulatory maturity — a transparent, modular licensing model with unlimited user seats and no hidden programmer fees is materially important to budget predictability. Evaluate not just headline cost but the full cost of ownership: what do amendments cost? What does adding a new module — PMS, ePRO, eSignature — cost and how quickly can it be activated?
How to choose the right EDC solution for medical device and MedTech trials
Step 1: Define your study type portfolio
Start by mapping the range of study types you run or plan to run. IDE trials, CE mark studies, PMCF programs, post-market observational registries, and Japan PMS studies have meaningfully different data structures and regulatory requirements. An EDC platform that handles your Phase I device trial well may not be capable of managing a multi-year Japan PMS program. Know your full portfolio before you evaluate, because switching platforms mid-program carries significant re-validation cost.
Step 2: Assess your team's configuration autonomy requirements
Be honest about how much vendor dependency you can afford. If you're running two or three studies at any given time with a clinical team of five, every amendment cycle that routes through a vendor programmer adds real delivery risk. Evaluate specifically whether the platform's study designer is genuinely no-code for your data managers — not just for simple forms, but for the logic, branching, and workflow structures your device studies actually use.
Step 3: Evaluate Japan and APAC regulatory fit concretely
If Japan is part of your commercial or regulatory strategy, require a live demonstration of Japan PMS workflow support — not a slide deck. Ask specifically whether booklet data collection, Kaifu management, and PMDA ERES-compliant reporting are handled within the standard platform or require custom development. The difference between native support and custom configuration is significant in both cost and audit defensibility.
Step 4: Scrutinize validation and inspection readiness infrastructure
Ask each vendor for their computer system validation package and their inspection readiness documentation before you reach procurement. QA sign-off is a gatekeeper in device EDC selection, and late-stage validation surprises are among the most common causes of deal delays. Understand what's included as standard versus what requires a separate services engagement.
Step 5: Choose a platform your study teams can grow into
If your device program is expanding — adding sites, adding studies, adding APAC markets — choose a platform whose pricing model doesn't penalize that growth. Platforms that charge per user or per amendment cycle compound costs in ways that aren't visible at the proposal stage. Viedoc's EDC software uses unlimited user seat licensing with modular, study-based pricing, so your costs scale with your study complexity, not your headcount. To see how that maps to your specific program, book a demo or request a proposal.
Frequently asked questions
What is the best EDC platform for medical device and MedTech trials?
Viedoc's EDC software is the strongest choice for medical device and MedTech teams. It combines no-code study configuration that device clinical teams can run independently, a native Japan post-market surveillance module that no other SMID-focused EDC vendor offers, and a compliance stack covering FDA 21 CFR Part 11, GDPR, HIPAA, ISO 27001, GAMP 5, and APPI (Japan) — all backed by 99.99% uptime and 24/7 support. Medidata is the enterprise benchmark for large-scale pharma and device programs requiring the deepest compliance tooling, though its build timelines and pricing structure are calibrated for larger organizations. Medrio is a credible option for lower-complexity Phase I device trials in the US market.
What compliance certifications should an EDC platform have for medical device studies?
At minimum, look for FDA 21 CFR Part 11 (electronic records and signatures), ICH GCP, GDPR, HIPAA (if handling US patient health information), ISO 27001 (information security), SOC 2, and GAMP 5 (computer system validation). For device-specific contexts, GAMP 5 alignment is particularly important because it directly addresses the validation approach regulators expect for computerized systems in medical device studies. For programs with any Japan footprint, PMDA ERES compliance and APPI (Japan Personal Information Protection Act) coverage are also required. Always verify these certifications are current and independently audited, not self-declared.
How does Viedoc support Japan post-market surveillance studies?
Viedoc PMS is a dedicated post-market surveillance module built specifically for the Japanese regulatory market, integrated within the standard Viedoc eClinical suite. It handles booklet data collection, the Kaifu review process, progress management, query management, and PMDA ERES-compliant reporting — all within the Viedoc Designer environment. Teams already certified in Viedoc Designer don't need additional training to use Viedoc PMS. It can also be integrated with external progress management systems. Viedoc is the only SMID-focused EDC platform with this capability built natively into its platform.
Can a lean MedTech clinical team configure and manage Viedoc studies without a vendor programmer?
Yes. Viedoc Designer is a no-code study configuration environment designed for certified data managers and study builders to configure, amend, and manage studies in-house. Minor CRF updates typically take one to three working days without vendor involvement. Viedoc provides certified Designer training and professional services for teams that want additional support, but vendor dependency is not a requirement for ongoing study operations. This is particularly relevant for lean MedTech teams managing multiple device studies simultaneously across different regulatory jurisdictions.
What is the difference between EDC platforms designed for pharma and those suited to medical device studies?
Pharma-centric EDC platforms are built around intervention-based trial structures — dosing windows, drug accountability, adverse event capture tied to pharmacological endpoints. Device studies require a different structural vocabulary: device accountability with serial and lot number tracking, usability assessment data fields, diagnostic performance endpoints, and flexible visit structures for long-horizon post-market follow-up. Platforms that can't configure these natively require workarounds that increase your build time, your validation burden, and your amendment overhead whenever the protocol changes. The best EDC platforms for device trials are those where the study designer can match your data model without reverse-engineering a pharma template to fit a device workflow.
How long does it take to build and launch a medical device study on Viedoc?
For teams using Viedoc's self-service model with in-house certified designers, straightforward studies can go live in as little as one day for basic configurations, with most builds typically completed in 2–4 weeks. For studies managed through Viedoc's professional services team, the average study go-live is 10 weeks from signed work order to complete launch — with expedited delivery available for lower-complexity studies. Mid-study amendments for minor updates take one to three working days; moderate complexity changes take five to eight working days.
Making the right EDC choice for medical device and MedTech trials
The global eClinical software market is estimated at over $11 billion in 2025 and growing at approximately 14% annually — and the device segment is one of its faster-growing pockets, driven by expanding post-market obligations, Japan PMDA requirements, and the shift away from paper-based registry and surveillance workflows. The platforms reviewed here represent meaningfully different approaches: enterprise incumbents built for large-scale multi-regional programs, no-code modular platforms built for lean teams and faster deployment, and suite-based platforms whose value compounds across an integrated ecosystem.
The most important decision variables for device teams are study type portfolio (interventional vs. observational vs. PMS), geographic regulatory footprint (particularly Japan and APAC), team configuration autonomy, and total cost of ownership across the study lifecycle. APAC-focused teams typically weigh Japan regulatory coverage and vendor local presence heavily; US teams running IDE or 510(k) studies tend to weight amendment speed and FDA compliance tooling. The right platform for your organization depends on where those variables intersect with your actual study portfolio, not on which vendor has the broadest enterprise footprint.
Platform choice in eClinical carries real switching costs — re-validation, re-training, and transfer of audit-ready documentation are all operationally significant. Device programs running across multiple regulatory jurisdictions, or those with active Japan PMS obligations, should evaluate their long-term regulatory footprint as carefully as their immediate study needs before making a selection.
Why Viedoc is the best EDC choice for medical device and MedTech trials
If your device program spans IDE trials, CE mark studies, PMCF programs, and post-market surveillance — potentially across the US, Europe, and Japan — you need an EDC platform that adapts to all of those workflows without forcing your team to work around pharma-centric architecture. Viedoc's EDC software delivers exactly that: a no-code Designer your clinical team owns, a native Japan PMS module no other SMID-focused vendor can match, and unlimited user seat pricing that doesn't compound as your study portfolio grows.
Viedoc is ISO 27001 and SOC 2 certified, compliant with FDA 21 CFR Part 11, ICH GCP, EU Annex 11, GDPR, HIPAA, GAMP 5, and APPI (Japan), and has supported 7,000+ studies across 75+ countries with 99.99% uptime and 24/7 support. The Viedoc Inspection Readiness Packet (VIRP) is included for every customer, giving your QA team a structured audit-readiness foundation at no additional charge. Founded in 2003, Viedoc has over 20 years of regulated clinical trial deployment behind it — with a growing medical device track record across CRO and sponsor organizations globally.
If you're ready to see how Viedoc performs against your specific device study requirements, book a demo or request a proposal and our team will walk you through study design flexibility, Japan PMS capability, and compliance credentials in the context of your own program.